Co-reporter:Raghuvir K. Arni;Nicolay Genov;Michaela Öhler;Jana Seifert;Martin von Bergen;Dessislava Georgieva
Journal of Proteome Research May 7, 2010 Volume 9(Issue 5) pp:2302-2316
Publication Date(Web):2017-2-22
DOI:10.1021/pr901042p
The snake venomic of Crotalus durissus terrificus was analyzed by 2-D and 1-D electrophoresis and subsequent MS/MS and enzymatic assays. The venomic of the South American rattlesnake comprises toxins from seven protein families: phospholipases A2, serine proteinases, ecto-5′-nucleotidases, metalloproteinases, nerve growth factors, phosphodiesterases, and glutaminyl cyclase. The venom toxin composition correlates with the clinical manifestation of the crotalinae snake bites and explains pathological effects of the venom such as neurotoxicity, systemic myonecrosis, hemostatic disorders, myoglobinuria, and acute renal failure. The vast majority of toxins are potentially involved in neurotoxicity, myotoxicity, and coagulopathy. The predominant venom components are neurotoxic phospholipases A2 and serine proteinases. The venom is a rich source of 5′-nucleotidases (7.8% of the identified toxins) inducing hemostatic disorders. Analysis of the venom protein composition provided a catalogue for secreted toxins. The venomic composition of Crotalus d. terrificus and venom gland transcriptome of the synonymous subspecies Crotalus d. collilineatus show differences in the occurrence of protein families and in the abundance of toxins. Some of the venom components identified by the proteomic analysis were not reported in the transcriptome of the Crotalus d. collilineatus venom gland. Enzymatic activities of the Crotalus d. terrificus venom were determined and correlated with the proteomic composition.Keywords: 2-D electrophoresis; Crotalus durissus terrificus; electrospray mass spectrometry; snake venomic;
Co-reporter:Robin SchubertArne Meyer, Daniela Baitan, Karsten Dierks, Markus Perbandt, Christian Betzel
Crystal Growth & Design 2017 Volume 17(Issue 3) pp:
Publication Date(Web):January 25, 2017
DOI:10.1021/acs.cgd.6b01826
Controlled navigation in the phase diagram of protein crystallization and probing by advanced Dynamic Light Scattering (DLS) technology provided new information and more insight on the early processes during the nucleation process. The observed hydrodynamic radius distribution pattern clearly reveals a two-step mechanism of nucleation and the occurrence of liquid dense protein clusters, which were verified by transmission electron microscopy. The growth kinetics of these protein clusters, forming distinct radii fractions, is analyzed in real time. Further, the data confirmed that critical nuclei show a distinctly different radius distribution than the liquid dense clusters. The data and results provide experimental evidence that during nucleation, a formation of distinct liquid clusters with high protein concentration occur prior to a transition to crystal nuclei by increasing the internal structural order of these clusters, subsequently.
Co-reporter:Chen-Yan Zhang, Yan Wang, Robin Schubert, Yue Liu, Meng-Yin Wang, Da Chen, Yun-Zhu Guo, Chen Dong, Hui-Meng Lu, Yong-Ming Liu, Zi-Qing Wu, Christian Betzel, and Da-Chuan Yin
Crystal Growth & Design 2016 Volume 16(Issue 2) pp:705-713
Publication Date(Web):January 5, 2016
DOI:10.1021/acs.cgd.5b01268
The successful crystallization of proteins is important because their molecular three-dimensional structures can be obtained through X-ray diffraction when in their crystalline form. Investigation of the crystallization process is beneficial for this purpose. We have reported that protein crystallization is sensitive to audible sound, which is commonly present but is often ignored. Here we investigate the effect of audible sound parameters, especially frequency, on a protein crystallization. We show a significant facilitation of protein crystallization using 5000 Hz audible sound, possible mechanism was also tried to be clarified. Suitably controlled audible sound can be beneficial for promoting protein crystallization. Therefore, audible sound can be used as a simple tool to promote protein crystallization. In addition, the processing of other types of materials may also be affected by audible sound.
Co-reporter:Arne Meyer;Karsten Dierks;Rana Hussein;Karl Brillet;Hevila Brognaro
Acta Crystallographica Section F 2015 Volume 71( Issue 1) pp:75-81
Publication Date(Web):
DOI:10.1107/S2053230X14027149
Detergents are widely used for the isolation and solubilization of membrane proteins to support crystallization and structure determination. Detergents are amphiphilic molecules that form micelles once the characteristic critical micelle concentration (CMC) is achieved and can solubilize membrane proteins by the formation of micelles around them. The results are presented of a study of micelle formation observed by in situ dynamic light-scattering (DLS) analyses performed on selected detergent solutions using a newly designed advanced hardware device. DLS was initially applied in situ to detergent samples with a total volume of approximately 2 µl. When measured with DLS, pure detergents show a monodisperse radial distribution in water at concentrations exceeding the CMC. A series of all-transn-alkyl-β-D-maltopyranosides, from n-hexyl to n-tetradecyl, were used in the investigations. The results obtained verify that the application of DLS in situ is capable of distinguishing differences in the hydrodynamic radii of micelles formed by detergents differing in length by only a single CH2 group in their aliphatic tails. Subsequently, DLS was applied to investigate the distribution of hydrodynamic radii of membrane proteins and selected water-insoluble proteins in presence of detergent micelles. The results confirm that stable protein–detergent complexes were prepared for (i) bacteriorhodopsin and (ii) FetA in complex with a ligand as examples of transmembrane proteins. A fusion of maltose-binding protein and the Duck hepatitis B virus X protein was added to this investigation as an example of a non-membrane-associated protein with low water solubility. The increased solubility of this protein in the presence of detergent could be monitored, as well as the progress of proteolytic cleavage to separate the fusion partners. This study demonstrates the potential of in situ DLS to optimize solutions of protein–detergent complexes for crystallization applications.
Co-reporter:Lars Redecke;Karol Nass;Daniel P. DePonte;Thomas A. White;Dirk Rehders;Anton Barty;Francesco Stellato;Mengning Liang;Thomas R.M. Barends;Sébastien Boutet;Garth J. Williams;Marc Messerschmidt;M. Marvin Seibert;Andrew Aquila;David Arnlund;Sasa Bajt;Torsten Barth;Michael J. Bogan;Carl Caleman;Tzu-Chiao Chao;R. Bruce Doak;Holger Fleckenstein;Matthias Frank;Raimund Fromme;Lorenzo Galli;Ingo Grotjohann;Mark S. Hunter;Linda C. Johansson;Stephan Kassemeyer;Gergely Katona;Richard A. Kirian;Rudolf Koopmann;Chris Kupitz;Lukas Lomb;Andrew V. Martin;Stefan Mogk;Richard Neutze;Robert L. Shoeman;Jan Steinbrener;Nicusor Timneanu;Dingjie Wang;Uwe Weierstall;Nadia A. Zatsepin;John C. H. Spence;Petra Fromme;Ilme Schlichting;Michael Duszenko;Henry N. Chapman
Science 2013 Vol 339(6116) pp:227-230
Publication Date(Web):11 Jan 2013
DOI:10.1126/science.1229663
Diffraction Before Destruction
A bottleneck in x-ray crystallography is the growth of well-ordered crystals large enough to obtain high-resolution diffraction data within an exposure that limits radiation damage. Serial femtosecond crystallography promises to overcome these constraints by using short intense pulses that out-run radiation damage. A stream of crystals is flowed across the free-electron beam and for each pulse, diffraction data is recorded from a single crystal before it is destroyed. Redecke et al. (p. 227, published online 29 November; see the Perspective by Helliwell) used this technique to determine the structure of an enzyme from Trypanosoma brucei, the parasite that causes sleeping sickness, from micron-sized crystals grown within insect cells. The structure shows how this enzyme, which is involved in degradation of host proteins, is natively inhibited prior to activation, which could help in the development of parasite-specific inhibitors.
Co-reporter:Dessislava Georgieva, Monika Coronado, Dominik Oberthür, Friedrich Buck, Deyan Duhalov, Raghuvir K. Arni and Christian Betzel
Molecular BioSystems 2012 vol. 8(Issue 5) pp:1405-1411
Publication Date(Web):23 Feb 2012
DOI:10.1039/C2MB05490F
Myotoxicity and membrane damage play a central role in the life-threatening effects of the viper envenomation. Myotoxins are an important part of the viper venomics. A Ser49 PLA2-like myotoxin from the venom of Vipera ammodytes meridionalis, the most venomous snake in Europe, was crystallized and its three-dimensional structure determined. The toxin is devoid of phospholipolytic activity. The structure demonstrates a formation of dimers. In the dimers functionally important peptide segments, located on the protein surface, point in the same direction which can strengthen the pharmacological effect. This supports the hypothesis about the physiological importance of the toxin oligomerization for the myotoxicity and membrane damage. The crystallographic model revealed that the structural determinants of myotoxicity (a positively charged C-terminal region and a hydrophobic knuckle) are fully exposed on the protein surface and accessible for interactions with target membranes. Distortion of the catalytic site region explains the absence of enzymatic activity. The structure reveals anion-binding sites which can be considered as possible sites of interactions of the toxin with a negatively charged membrane surface. The high structural similarity of the Ser49 myotoxin and Asp49 PLA2 from the same venom suggests an evolutionary relationship: probably, the Ser49 myotoxin is a product of evolution of the catalytically active phospholipase A2. The first toxin lost the enzymatic activity which is not necessary for the myotoxicity but preserved the cytotoxicity and membrane damaging activity as important components of the venom toxicity.
Co-reporter:Dessislava Georgieva, Jana Seifert, Michaela Öhler, Martin von Bergen, Patrick Spencer, Raghuvir K. Arni, Nicolay Genov, and Christian Betzel
Journal of Proteome Research 2011 Volume 10(Issue 5) pp:2440-2464
Publication Date(Web):2017-2-22
DOI:10.1021/pr101248e
The venom composition of Pseudechis australis, a widely distributed in Australia reptile, was analyzed by 2-DE and mass spectrometric analysis. In total, 102 protein spots were identified as venom toxins. The gel is dominated by horizontal trains of spots with identical or very similar molecular masses but differing in the pI values. This suggests possible post-translational modifications of toxins, changing their electrostatic charge. The results demonstrate a highly specialized biosynthesis of toxins destroying the hemostasis (P−III metalloproteases, SVMPs), antimicrobial proteins (l-amino acid oxidases, LAAOs, and transferrin-like proteins, TFLPs), and myotoxins (phospholipase A2s, PLA2s). The three transferrin isoforms of the Australian P. australis (Elapidae snake) venom are highly homologous to the body transferrin of the African Lamprophis fuliginosus (Colubridae), an indication for the recruitment of body transferrin. The venomic composition suggests an adaptation for a defense against microbial pathogens from the prey. Transferrins have not previously been reported as components of elapid or other snake venoms. Ecto-5′-nucleotidases (5′-NTDs), nerve growth factors (VNGFs), and a serine proteinase inhibitor (SPI) were also identified. The venom composition and enzymatic activities explain the clinical manifestation of the king brown snakebite. The results can be used for medical, scientific, and biotechnological purposes.
Co-reporter:Aisha Munawar, Maria Trusch, Dessislava Georgieva, Patrick Spencer, Violette Frochaux, Sönke Harder, Raghuvir K. Arni, Deyan Duhalov, Nicolay Genov, Hartmut Schlüter and Christian Betzel
Molecular BioSystems 2011 vol. 7(Issue 12) pp:3298-3307
Publication Date(Web):29 Sep 2011
DOI:10.1039/C1MB05309D
Snake venom peptidomes are valuable sources of pharmacologically active compounds. We analyzed the peptidic fractions (peptides with molecular masses < 10000 Da) of venoms of Vipera ammodytes meridionalis (Viperinae), the most toxic snake in Europe, and Bothrops jararacussu (Crotalinae), an extremely poisonous snake of South America. Liquid chromatography/mass spectrometry (LC/MS), direct infusion electrospray mass spectrometry (ESI-MS) and matrix-assisted desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS) were applied to characterize the peptides of both snake venoms. 32 bradykinin-potentiating peptides (BPPs) were identified in the Crotalinae venom and their sequences determined. 3 metalloproteinase inhibitors, 10 BPPs and a Kunitz-type inhibitor were observed in the Viperinae venom peptidome. Variability in the C-terminus of homologous BPPs was observed, which can influence the pharmacological effects. The data obtained so far show a subfamily specificity of the venom peptidome in the Viperidae family: BPPs are the major peptide component of the Crotalinae venom peptidome lacking Kunitz-type inhibitors (with one exception) while the Viperinae venom, in addition to BPPs, can contain peptides of the bovine pancreatic trypsin inhibitor family. We found indications for a post-translational phosphorylation of serine residues in Bothrops jararacussu venom BPP (QGLPPGPPIP), which could be a regulatory mechanism in their interactions with ACE, and might influence the hypotensive effect. Homology between venom BPPs from Viperidae snakes and venom natriuretic peptide precursors from Elapidae snakes suggests a structural similarity between the respective peptides from the peptidomes of both snake families. The results demonstrate that the venoms of both snakes are rich sources of peptides influencing important physiological systems such as blood pressure regulation and hemostasis. The data can be used for pharmacological and medical applications.
Co-reporter:Michaela Öhler, Dessislava Georgieva, Jana Seifert, Martin von Bergen, Raghuvir K. Arni, Nicolay Genov and Christian Betzel
Journal of Proteome Research 2010 Volume 9(Issue 5) pp:2422-2437
Publication Date(Web):2017-2-22
DOI:10.1021/pr901128x
The venom proteome of Bothrops alternatus, a venomous snake widespread in South America, was analyzed by 2-D electrophoresis followed by mass spectrometric analysis and determination of enzymatic activities. The venomic composition revealed that metallo- and serine proteinases play primary roles in the pathogenesis of the envenomation by this pitviper. The identified 100 venom components with molecular masses from 10 to 100 kDa belong to six protein families: metalloproteinases, serine/thrombin-like proteinases, phospholipases A2, L-amino acid oxidases, disintegrins and thrombin inhibitors. Metalloproteinases predominate and belong exclusively to the P-III class including the most potent hemorrhagic toxins. They represent 50% of all identified proteins. Two isoforms were identified: homologous to jararhagin, a hemorrhagic toxin, and to beritractivase, a nonhemorrhagic and pro-coagulant metalloproteinase. The B. alternatus venom is a rich source of proteins influencing the blood coagulation system with a potential for medical application. The isoelectric points of the components are distributed in the acidic pH range (the pI values are between 4 and 7) and no basic proteins were detected.
Co-reporter:Dessislava Georgieva, Kerstin Greunke and Christian Betzel
Molecular BioSystems 2010 vol. 6(Issue 6) pp:1056-1060
Publication Date(Web):17 Mar 2010
DOI:10.1039/B923127G
Api m 7 is one of the major protease allergens of the honeybee venom. It consists of a serine protease-like (SPL) and a CUB domain. The knowledge about the structure and function of Api m 7 is limited mainly to its amino acid sequence. Three-dimensional models of the two structural domains were constructed using their amino acid sequences and the crystallographic coordinates of prophenoloxidase-activating factor (PPAF-II) as a template for the SPL domain and the coordinates of porcine spermadhesin PSP-II for the CUB domain. The structural organization of Api m 7 suggests that the CUB domain is involved in interactions with natural substrates while the SPL domain probably activates zymogens. IgE epitopes and antigenic sites were predicted. Api m 7 shows structural and functional similarity to the members of the PPAF-II family. Possible substrates, function and evolution of the enzyme are discussed in the paper.


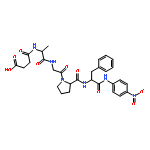
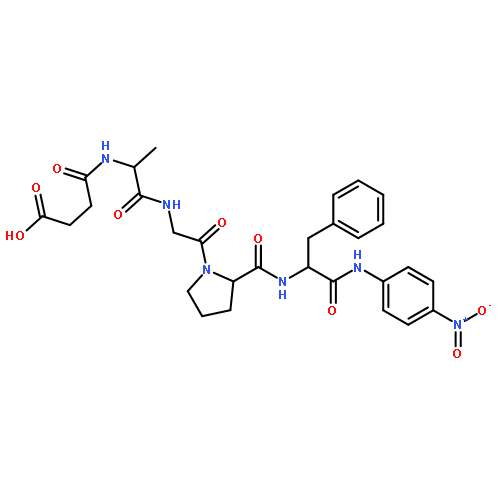





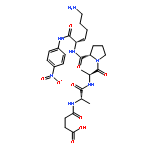
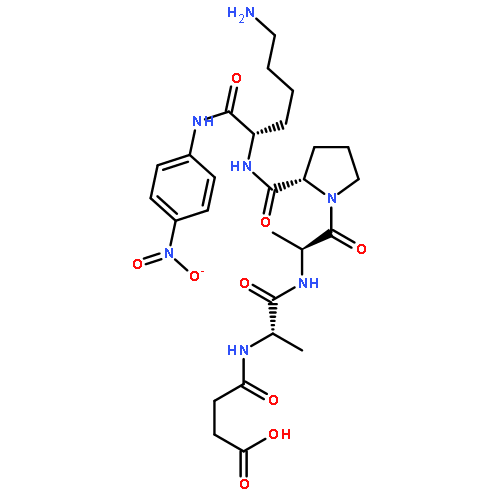
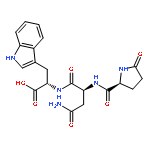
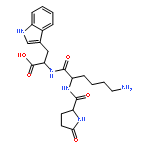
![L-Tryptophan, N-[N2-(5-oxo-L-prolyl)-L-arginyl]-](http://img.cochemist.com/ccimg/98400/98385-51-4.png)
![L-Tryptophan, N-[N2-(5-oxo-L-prolyl)-L-arginyl]-](http://img.cochemist.com/ccimg/98400/98385-51-4_b.png)
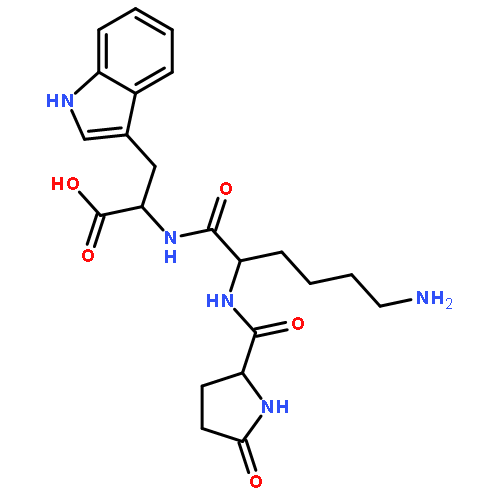




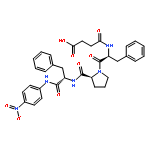

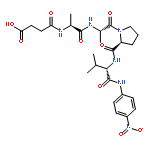
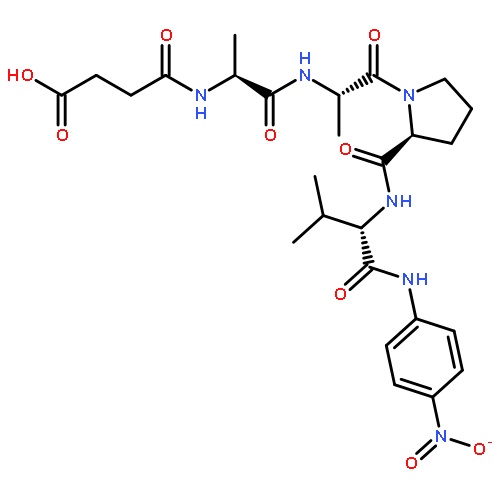

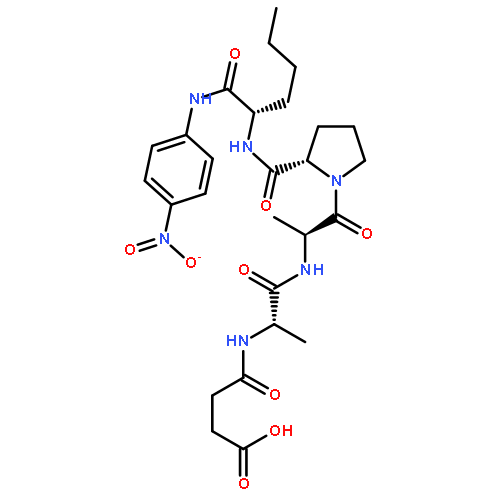






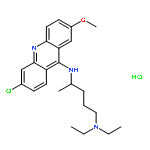
![N-(3-carboxypropanoyl)-L-alanyl-N-{(2S)-2-[(4-nitrophenyl)amino]-3-phenylpropanoyl}-L-alaninamide](http://img.cochemist.com/ccimg/61100/61043-53-6.png)
![N-(3-carboxypropanoyl)-L-alanyl-N-{(2S)-2-[(4-nitrophenyl)amino]-3-phenylpropanoyl}-L-alaninamide](http://img.cochemist.com/ccimg/61100/61043-53-6_b.png)