Co-reporter:Maykon Lima Souza, Anthony W. DeMartino, and Peter C. Ford
ACS Omega October 2017? Volume 2(Issue 10) pp:6535-6535
Publication Date(Web):October 9, 2017
DOI:10.1021/acsomega.7b01206
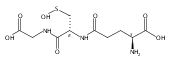
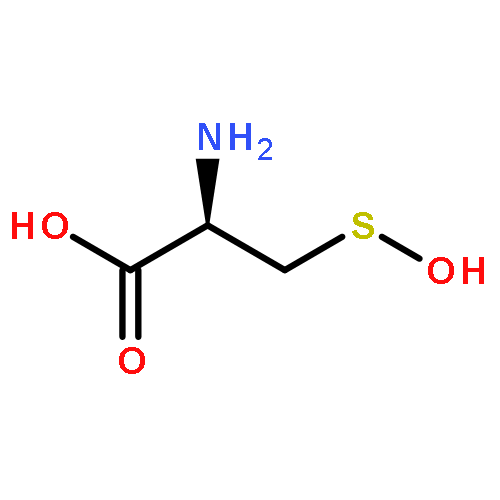
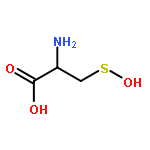
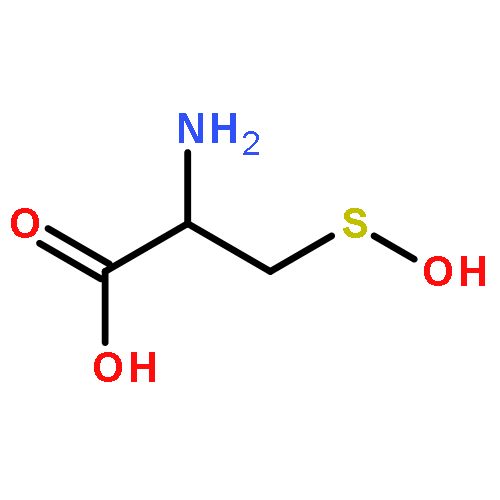
Carbon disulfide is an environmental toxin, but there are suggestions in the literature that it may also have regulatory and/or therapeutic roles in mammalian physiology. Thiols or thiolates would be likely biological targets for an electrophile, such as CS2, and in this context, the present study examines the dynamics of CS2 reactions with various thiols (RSH) in physiologically relevant near-neutral aqueous media to form the respective trithiocarbonate anions (TTC–, also known as “thioxanthate anions”). The rates of TTC– formation are markedly pH-dependent, indicating that the reactive form of RSH is the conjugate base RS–. The rates of the reverse reaction, that is, decay of TTC– anions to release CS2, is pH-independent, with rates roughly antiparallel to the basicities of the RS– conjugate base. These observations indicate that the rate-limiting step of decay is simple CS2 dissociation from RS–, and according to microscopic reversibility, the transition state of TTC– formation would be simple addition of the RS– nucleophile to the CS2 electrophile. At pH 7.4 and 37 °C, cysteine and glutathione react with CS2 at a similar rate but the trithiocarbonate product undergoes a slow cyclization to give 2-thiothiazolidine-4-carboxylic acid. The potential biological relevance of these observations is briefly discussed.Topics: Biological and Medicinal chemistry; Enthalpy; Enthalpy; Entropy; Entropy; Quantum mechanical methods; Quantum mechanical methods; Reaction kinetics; Reaction kinetics; Spectra;
Co-reporter:Anthony W. DeMartino;David F. Zigler;Jon M. Fukuto
Chemical Society Reviews 2017 vol. 46(Issue 1) pp:21-39
Publication Date(Web):2017/01/03
DOI:10.1039/C6CS00585C
The overview presented here has the goal of examining whether carbon disulfide (CS2) may play a role as an endogenously generated bioregulator and/or has therapeutic value. The neuro- and reproductive system toxicity of CS2 has been documented from its long-term use in the viscose rayon industry. CS2 is also used in the production of dithiocarbamates (DTCs), which are potent fungicides and pesticides, thus raising concern that CS2 may be an environmental toxin. However, DTCs also have recognized medicinal use in the treatment of heavy metal poisonings as well as having potency for reducing inflammation. Three known small molecule bioregulators (SMBs) nitric oxide, carbon monoxide, and hydrogen sulfide were initially viewed as environmental toxins. Yet each is now recognized as having intricate, though not fully elucidated, biological functions at concentration regimes far lower than the toxic doses. The literature also implies that the mammalian chemical biology of CS2 has broader implications from inflammatory states to the gut microbiome. On these bases, we suggest that the very nature of CS2 poisoning may be related to interrupting or overwhelming relevant regulatory or signaling process(es), much like other SMBs.
Co-reporter:Anthony W. DeMartino;Maykon Lima Souza
Chemical Science (2010-Present) 2017 vol. 8(Issue 10) pp:7186-7196
Publication Date(Web):2017/09/25
DOI:10.1039/C7SC02727C
We describe the kinetics of the formation and decay of a series of dithiocarbamates under physiological conditions. The goal is to provide a toolbox of compounds that release CS2 by well-defined kinetics in such media. Carbon disulfide is a known environmental toxin, but there is fragmentary evidence suggesting that CS2 may have bioregulatory and/or therapeutic roles in mammalian biology. Further investigation of such roles will require methodologies for controlled delivery of this bioactive small molecule to specific targets. Reported here are mechanistic and computational studies of CS2 release from a series of dithiocarbamate anions (DTCs), where R2N represents several different secondary amido groups. The various DTCs under physiologically relevant conditions show a tremendous range of reactivities toward CS2 dissociation with decay lifetimes ranging from ∼2 s for imidazolidyldithiocarbamate (ImDTC−) to ∼300 s for diisopropyldithiocarbamate (DIDTC−) to >24 h for pyrrolidinyldithiocarbamate (PDTC−) in pH 7.4 phosphate buffer solution at 37 °C. Thus, by making the correct choice of these tools, one can adjust the flux of CS2 in a biological experiment, while the least reactive DTCs could serve as controls for evaluating the potential effects of the dithiocarbamate functionality itself. Kinetics studies and density functional calculations are used to probe the mechanism of DTC− decay. In each case, the rate of CS2 dissociation is acid dependent; however, the DFT studies point to a mechanistic pathway for ImDTC− that is different than those for DIDTC−. The role of general acid catalysis is also briefly probed.
Co-reporter:Tigran S. Kurtikyan, Astghik A. Hovhannisyan, and Peter C. Ford
Inorganic Chemistry 2016 Volume 55(Issue 19) pp:9517-9520
Publication Date(Web):September 19, 2016
DOI:10.1021/acs.inorgchem.6b01744
Low-temperature in situ Fourier transform infrared and UV–vis measurements show that trimethylphosphine (PMe3) reacts with microporous layers of FeII(TTP)(NO) (TTP = meso-tetra-p-tolylporphyrinato dianion; NO = nitric oxide) to form the previously unknown six-coordinate complex FeII(TTP)(PMe3)(NO). Upon warming this compound to room temperature in the presence of excess phosphine, the NO ligand is completely replaced by phosphine, resulting in formation of the bis(trimethylphosphine) complex FeII(TTP)(PMe3)2. Simultaneously, the NO released oxidizes free PMe3 to the corresponding phosphine oxide (OPMe3) with concomitant formation of nitrous oxide (N2O).
Co-reporter:Christopher M. Bernt, Giovanni Bottari, Jacob A. Barrett, Susannah L. Scott, Katalin Barta and Peter C. Ford
Catalysis Science & Technology 2016 vol. 6(Issue 9) pp:2984-2994
Publication Date(Web):27 Nov 2015
DOI:10.1039/C5CY01555C
Copper-doped porous metal oxides catalyze the one-pot disassembly of biomass-derived lignin via C–O bond hydrogenolysis and hydrodeoxygenation in supercritical methanol. This catalytic system cleanly converts lignin as well as lignocellulose composites, such as sawdust, to organic liquids with little or no formation of intractable tars or chars. However, this catalyst based on Earth-abundant components also catalyzes less desirable aromatic ring hydrogenations and various methylations that contribute to the diversity of products. In this context, we undertook a quantitative experimental and computational evaluation of model reactions relevant to the reductive disassembly of lignin by this catalyst system in order to determine quantitatively the rates of desirable and less desirable chemical steps that define the overall product selectivities. Global fitting analysis methods were used to map the temporal evolution of key intermediates and products and to elucidate networks that provide guidelines regarding the eventual fates of reactive intermediates in this catalysis system. Phenolic compounds display multiple reaction pathways, but substrates such as benzene, toluene, and alkyl- and alkoxy-substituted aromatics are considerably more stable under these conditions. These results indicate that modifying this catalytic system in a way that controls and channels the reactivity of phenolic intermediates should improve selectivity toward producing valuable aromatic chemicals from biomass-derived lignin. To this end we demonstrate that the O-methylating agent dimethyl carbonate can intercept the phenol intermediate formed from hydrogenolysis of the model compound benzyl phenyl ether. Trapping the phenol as anisole thus gave much higher selectivity towards aromatic products.
Co-reporter:Madeline A. Weber, Peter C. Ford
Journal of Molecular Catalysis A: Chemical 2016 Volume 416() pp:81-87
Publication Date(Web):15 May 2016
DOI:10.1016/j.molcata.2016.02.018
•Dehydrogenation of 1,2-diols leads overwhelmingly to α-hydroxyketone (α-HK) as product.•Tandem decarbonylation with Rh catalyst indicates α-hydroxyaldehydes (α-HAs) are likely intermediates•α-HA transformation to more stable α-HK is by hydrogenation/rehydrogenation steps.Described are studies of the dehydrogenation of 1,2- and 1,3-diols in homogenous solutions catalyzed by {[2,5-diphenyl-3,4-ditoluyl-(η5-C4CO)]2H}Ru2(CO)4(μ-H) (otherwise known as the Casey/Shvo catalyst). Both in the presence and absence of a dihydrogen acceptor, these reactions led to the analogous α-hydroxyketone as the only organic product. Isotopic labeling studies indicate that this product arises from reversible dehydrogenation/hydrogenation reactions, resulting in formation of the thermodynamically favored α-hydroxyketone. When this catalytic dehydrogenation was carried out in the presence of the rhodium decarbonylation catalyst Rh(dppp)2Cl (dppp = 1,3-bis(diphenylphosphino)propane), modest amounts of carbon monoxide result, suggesting that the dehydrogenation does generate at least some aldehydes that are intercepted by this catalyst. However, the efficiency of the latter reaction is poor.
Co-reporter:Jacob A. Barrett, Yu Gao, Christopher M. Bernt, Megan Chui, Anthony T. Tran, Marcus B. Foston, and Peter. C. Ford
ACS Sustainable Chemistry & Engineering 2016 Volume 4(Issue 12) pp:
Publication Date(Web):September 15, 2016
DOI:10.1021/acssuschemeng.6b01827
The selective conversion of lignin into aromatic compounds has the potential to serve as a “green” alternative to the production of petrochemical aromatics. Herein, we evaluate the addition of dimethyl carbonate (DMC) to a biomass conversion system that uses a Cu-doped porous metal oxide (Cu20PMO) catalyst in supercritical methanol (sc-MeOH) to disassemble lignin with little to no char formation. While Cu20PMO catalyzes C–O hydrogenolysis of aryl–ether bonds linking lignin monomers, it also catalyzes arene methylation and hydrogenation, leading to product proliferation. The MeOH/DMC co-solvent system significantly suppresses arene hydrogenation of the phenolic intermediates responsible for much of the undesirable product diversity via O-methylation of phenolic −OH groups to form more stable aryl-OCH3 species. Consequently, product proliferation was greatly reduced and aromatic yields greatly enhanced with lignin models, 2-methoxy-4-propylphenol, benzyl phenyl ether, and 2-phenoxy-1-phenylethan-1-ol. In addition, organosolv poplar lignin (OPL) was examined as a substrate in the MeOH/DMC co-solvent system. The products were characterized by nuclear magnetic resonance spectroscopy (31P, 13C, and 2D 1H–13C NMR) and gas chromatography–mass spectrometry techniques. The co-solvent system demonstrated enhanced yields of aromatic products.Keywords: Dimethyl carbonate; Heterogeneous catalysis; Hydrotalcite; Lignin; Porous metal oxides; Supercritical methanol;
Co-reporter:Elizabeth S. Levy, Demosthenes P. Morales, John V. Garcia, Norbert O. Reich and Peter C. Ford
Chemical Communications 2015 vol. 51(Issue 100) pp:17692-17695
Publication Date(Web):16 Oct 2015
DOI:10.1039/C5CC07989F
We demonstrate modulation of nitric oxide release in solution and in human prostate cancer cells from a thiol functionalized cupferron (TCF) absorbed on hollow gold nanoshells (HGNs) using near-infrared (NIR) light. NO release from the TCF–HGN conjugates occurs through localized surface heating due to NIR excitation of the surface plasmon. Specific HGN targeting is achieved through cell surface directed peptides, and excitation with tissue penetrating NIR light provides unprecedented spatio-temporal control of NO delivery to biological targets.
Co-reporter:Agustin E. Pierri, Po-Ju Huang, John V. Garcia, James G. Stanfill, Megan Chui, Guang Wu, Nanfeng Zheng and Peter C. Ford
Chemical Communications 2015 vol. 51(Issue 11) pp:2072-2075
Publication Date(Web):16 Dec 2014
DOI:10.1039/C4CC06766E
A water-soluble nanocarrier for a photo-activated CO releasing moiety (photoCORM) that can be triggered with NIR excitation is described. This has an upconversion nanoparticle core encapsulated by an amphiphilic polymer imparting both water solubility and a hydrophobic interior containing the photoCORM trans-Mn(bpy)(PPh3)2(CO)2. Such an ensemble offers a unique strategy for CO delivery to biological targets.
Co-reporter:Katalin Barta and Peter C. Ford
Accounts of Chemical Research 2014 Volume 47(Issue 5) pp:1503
Publication Date(Web):April 18, 2014
DOI:10.1021/ar4002894
This Account outlines recent efforts in our laboratories addressing a fundamental challenge of sustainability chemistry, the effective utilization of biomass for production of chemicals and fuels. Efficient methods for converting renewable biomass solids to chemicals and liquid fuels would reduce society’s dependence on nonrenewable petroleum resources while easing the atmospheric carbon dioxide burden. The major nonfood component of biomass is lignocellulose, a matrix of the biopolymers cellulose, hemicellulose, and lignin. New approaches are needed to effect facile conversion of lignocellulose solids to liquid fuels and to other chemical precursors without the formation of intractable side products and with sufficient specificity to give economically sustainable product streams.We have devised a novel catalytic system whereby the renewable feedstocks cellulose, organosolv lignin, and even lignocellulose composites such as sawdust are transformed into organic liquids. The reaction medium is supercritical methanol (sc-MeOH), while the catalyst is a copper-doped porous metal oxide (PMO) prepared from inexpensive, Earth-abundant starting materials. This transformation occurs in a single stage reactor operating at 300–320 °C and 160–220 bar. The reducing equivalents for these transformations are derived by the reforming of MeOH (to H2 and CO), which thereby serves as a “liquid syngas” in the present case. Water generated by deoxygenation processes is quickly removed by the water–gas shift reaction. The Cu-doped PMO serves multiple purposes, catalyzing substrate hydrogenolysis and hydrogenation as well as the methanol reforming and shift reactions. This one-pot “UCSB process” is quantitative, giving little or no biochar residual.Provided is an overview of these catalysis studies beginning with reactions of the model compound dihydrobenzofuran that help define the key processes occurring. The initial step is phenyl–ether bond hydrogenolysis, and this is followed by aromatic ring hydrogenation. The complete catalytic disassembly of the more complex organosolv lignin to monomeric units, largely propyl-cyclohexanol derivatives is then described. Operational indices based on 1H NMR analysis are also presented that facilitate holistic evaluation of these product streams that within several hours consist largely of propyl-cyclohexanol derivatives. Lastly, we describe the application of this methodology with several types of wood (pine sawdust, etc.) and with cellulose fibers. The product distribution, albeit still complex, displays unprecedented selectivity toward the production of aliphatic alcohols and methylated derivatives thereof. These observations clearly indicate that the Cu-doped solid metal oxide catalyst combined with sc-MeOH is capable of breaking down the complex biomass derived substrates to markedly deoxygenated monomeric units with increased hydrogen content. Possible implementations of this promising system on a larger scale are discussed.
Co-reporter:José Clayston Melo Pereira; Alexei V. Iretskii; Rui-Min Han
Journal of the American Chemical Society 2014 Volume 137(Issue 1) pp:328-336
Publication Date(Web):December 5, 2014
DOI:10.1021/ja510393q
Kinetics studies provide mechanistic insight regarding the formation of dinitrosyl iron complexes (DNICs) now viewed as playing important roles in the mammalian chemical biology of the ubiquitous bioregulator nitric oxide (NO). Reactions in deaerated aqueous solutions containing FeSO4, cysteine (CysSH), and NO demonstrate that both the rates and the outcomes are markedly pH dependent. The dinuclear DNIC Fe2(μ-CysS)2(NO)4, a Roussin’s red salt ester (Cys-RSE), is formed at pH 5.0 as well as at lower concentrations of cysteine in neutral pH solutions. The mononuclear DNIC Fe(NO)2(CysS)2– (Cys-DNIC) is produced from the same three components at pH 10.0 and at higher cysteine concentrations at neutral pH. The kinetics studies suggest that both Cys-RSE and Cys-DNIC are formed via a common intermediate Fe(NO)(CysS)2–. Cys-DNIC and Cys-RSE interconvert, and the rates of this process depend on the cysteine concentration and on the pH. Flash photolysis of the Cys-RSE formed from Fe(II)/NO/cysteine mixtures in anaerobic pH 5.0 solution led to reversible NO dissociation and a rapid, second-order back reaction with a rate constant kNO = 6.9 × 107 M–1 s–1. In contrast, photolysis of the mononuclear-DNIC species Cys-DNIC formed from Fe(II)/NO/cysteine mixtures in anaerobic pH 10.0 solution did not labilize NO but instead apparently led to release of the CysS• radical. These studies illustrate the complicated reaction dynamics interconnecting the DNIC species and offer a mechanistic model for the key steps leading to these non-heme iron nitrosyl complexes.
Co-reporter:T. S. Kurtikyan, V. A. Hayrapetyan, M. M. Mehrabyan, and P. C. Ford
Inorganic Chemistry 2014 Volume 53(Issue 22) pp:11948-11959
Publication Date(Web):November 4, 2014
DOI:10.1021/ic5014329
Reaction of small increments of NO2 gas with sublimed amorphous layers of MnII(TPP) (TPP = meso-tetra-phenylporphyrinato dianion) in a vacuum cryostat leads to formation of the 5-coordinate monodentate nitrato complex MnIII(TPP)(η1-ONO2) (II). This transformation proceeds through the two distinct steps with initial formation of the five coordinate O-nitrito complex MnIII(TPP)(η1-ONO) (I) as demonstrated by the electronic absorption spectra and by FTIR spectra using differently labeled nitrogen dioxide. A plausible mechanism for the second stage of reaction is offered based on the spectral changes observed upon subsequent interaction of 15NO2 and NO2 with the layered Mn(TPP). Low-temperature interaction of I and II with the vapors of various ligands L (L = O-, S-, and N-donors) leads to formation of the 6-coordinate O-nitrito MnIII(TPP)(L)(η1-ONO) and monodentate nitrato MnIII(TPP)(L)(η1-ONO2) complexes, respectively. Formation of the 6-coordinate O-nitrito complex is accompanied by the shifts of the ν(N═O) band to lower frequency and of the ν(N–O) band to higher frequency. The frequency difference between these bands Δν = ν(N═O) – ν(N–O) is a function of L and is smaller for the stronger bases. Reaction of excess NH3 with I leads to formation of Mn(TPP)(NH3)(η1-ONO) and of the cation [Mn(TPP)(NH3)2]+ plus ionic nitrite. The nitrito complexes are relatively unstable, but several of the nitrato species can be observed in the solid state at room temperature. For example, the tetrahydrofuran complex Mn(TPP)(THF)(η1-ONO2) is stable in the presence of THF vapors (∼5 mm), but it loses this ligand upon high vacuum pumping at RT. When L = dimethylsulfide (DMS), the nitrato complex is stable only to ∼−30 °C. Reactions of II with the N-donor ligands NH3, pyridine, or 1-methylimidazole are more complex. With these ligands, the nitrato complexes MnIII(TPP)(L)(η1-ONO2) and the cationic complexes [Mn(TPP)(L)2]+ coexist in the layer at room temperature, the latter formed as a result of NO3– displacement when L is in excess.
Co-reporter:Lilian Pereira Franco, Simone Aparecida Cicillini, Juliana Cristina Biazzotto, Marco A. Schiavon, Alexander Mikhailovsky, Peter Burks, John Garcia, Peter C. Ford, and Roberto Santana da Silva
The Journal of Physical Chemistry A 2014 Volume 118(Issue 51) pp:12184-12191
Publication Date(Web):November 18, 2014
DOI:10.1021/jp5111218
We describe the use of cadmium telluride quantum dots (CdTe QDs) as antennas for the photosensitization of nitric oxide release from a ruthenium nitrosyl complex with visible light excitation. The CdTe QDs were capped with mercaptopropionic acid to make them water-soluble, and the ruthenium nitrosyl complex was cis-[Ru(NO)(4-ampy)(bpy)2]3+ (Ru–NO; bpy is 2,2′-bipyridine, and 4-ampy is 4-aminopyridine). Solutions of these two components demonstrated concentration-dependent quenching of the QD photoluminescence (PL) as well as photoinduced release of NO from Ru–NO when irradiated by 530 nm light. A NO release enhancement of ∼8 times resulting from this association was observed under longer wavelength excitation in visible light range. The dynamics of the quenching determined by both PL and transient absorption measurements were probed by ultrafast flash photolysis. A charge transfer mechanism is proposed to explain the quenching of the QD excited states as well as the photosensitized release of NO from Ru–NO.
Co-reporter:Peter T. Burks ; John V. Garcia ; Ricardo GonzalezIrias ; Jason T. Tillman ; Mutong Niu ; Alexander A. Mikhailovsky ; Jinping Zhang ; Fan Zhang
Journal of the American Chemical Society 2013 Volume 135(Issue 48) pp:18145-18152
Publication Date(Web):November 18, 2013
DOI:10.1021/ja408516w
Novel materials for the phototherapeutic release of the bioregulator nitric oxide (nitrogen monoxide) are described. Also reported is a method for scanning these materials with a focused NIR beam to induce photouncaging while minimizing damage from local heating. The new materials consist of poly(dimethylsiloxane) composites with near-infrared-to-visible upconverting nanoparticles (UCNPs) that are cast into a biocompatible polymer disk (PD). These PDs are then impregnated with the photochemical nitric oxide precursor Roussin’s black salt (RBS) to give UCNP_RBS_PD devices that generate NO when irradiated with 980 nm light. When the UCNP_RBS_PD composites were irradiated with NIR light through filters composed of porcine tissue, physiologically relevant NO concentrations were released, thus demonstrating the potential of such devices for minimally invasive phototherapeutic applications.
Co-reporter:Christopher M. Bernt ; Peter T. Burks ; Anthony W. DeMartino ; Agustin E. Pierri ; Elizabeth S. Levy ; David F. Zigler
Journal of the American Chemical Society 2013 Volume 136(Issue 6) pp:2192-2195
Publication Date(Web):October 23, 2013
DOI:10.1021/ja4083599
Carbon disulfide, a potentially therapeutic small molecule, is generated via oxidative cleavage of 1,1-dithiooxalate (DTO) photosensitized by CdSe quantum dots (QDs). Irradiation of DTO–QD conjugates leads to λirr independent photooxidation with a quantum yield of ∼4% in aerated pH 9 buffer solution that drops sharply in deaerated solution. Excess DTO is similarly decomposed, indicating labile exchange at the QD surfaces and a photocatalytic cycle. Analogous photoreaction occurs with the O-tert-butyl ester tBuDTO in nonaqueous media. We propose that oxidation is initiated by hole transfer from photoexcited QD to surface DTO and that these substrates are a promising class of photocleavable ligands for modifying QD surface coordination.
Co-reporter:Julie L. Heinecke ; Chosu Khin ; Jose Clayston Melo Pereira ; Sebastián A. Suárez ; Alexei V. Iretskii ; Fabio Doctorovich
Journal of the American Chemical Society 2013 Volume 135(Issue 10) pp:4007-4017
Publication Date(Web):February 19, 2013
DOI:10.1021/ja312092x
The water-soluble ferriheme model FeIII(TPPS) mediates oxygen atom transfer from inorganic nitrite to a water-soluble phosphine (tppts), dimethyl sulfide, and the biological thiols cysteine (CysSH) and glutathione (GSH). The products with the latter reductant are the respective sulfenic acids CysS(O)H and GS(O)H, although these reactive intermediates are rapidly trapped by reaction with excess thiol. The nitrosyl complex FeII(TPPS)(NO) is the dominant iron species while excess substrate is present. However, in slightly acidic media (pH ≈ 6), the system does not terminate at this very stable ferrous nitrosyl. Instead, it displays a matrix of redox transformations linking spontaneous regeneration of FeIII(TPPS) to the formation of both N2O and NO. Electrochemical sensor and trapping experiments demonstrate that HNO (nitroxyl) is formed, at least when tppts is the reductant. HNO is the likely predecessor of the N2O. A key pathway to NO formation is nitrite reduction by FeII(TPPS), and the kinetics of this iron-mediated transformation are described. Given that inorganic nitrite has protective roles during ischemia/reperfusion (I/R) injury to organs, attributed in part to NO formation, and that HNO may also reduce net damage from I/R, the present studies are relevant to potential mechanisms of such nitrite protection.
Co-reporter:Arsen S. Azizyan, Tigran S. Kurtikyan, Garik G. Martirosyan, and Peter C. Ford
Inorganic Chemistry 2013 Volume 52(Issue 9) pp:5201-5205
Publication Date(Web):April 10, 2013
DOI:10.1021/ic400102q
Interaction of NO (15NO) with amorphous layers of Ru(II) carbonyl porphyrin (Ru(TPP)(CO), TPP2- = meso-tetraphenylporphyrinato dianion) was monitored by FTIR spectroscopy from 80 K to room temperature. An intermediate spectrally characterized at very low temperatures (110 K) with ν(CO) at 2001 cm–1 and ν(NO) at 1810 cm–1 (1777 cm–1 for 15NO isotopomer) was readily assigned to the mixed carbonyl–nitrosyl complex Ru(TPP)(CO)(NO), which is the logical precursor to CO labilization. Remarkably, Ru(TPP)-mediated disproportionation of NO is seen even at 110 K, an indication of how facile this reaction is. By varying the quantity of supplied NO, it was also demonstrated that the key intermediate responsible for NO disproportionation is the dinitrosyl complex Ru(TPP)(NO)2, supporting the conclusion previously made from solution experiments.
Co-reporter:G.G. Martirosyan, T.S. Kurtikyan, A.S. Azizyan, A.V. Iretskii, P.C. Ford
Journal of Inorganic Biochemistry 2013 Volume 121() pp:129-133
Publication Date(Web):April 2013
DOI:10.1016/j.jinorgbio.2012.12.017
The interaction of the S- and O-donor ligands tetrahydrothiophen (THT) and tetrahydrofuran (THF) with the ferrous nitrosyl complex Fe(TTP)(NO) (TTP2 − is meso-tetra-p-tolyl-porphyrinatodianion) was studied at various temperatures both in solid state and solution using electronic and infrared absorption spectroscopy. Upon addition of these ligands to a cryostat containing sublimed layers of Fe(TTP)(NO), no complex formation was detected at room temperature. However, upon lowering the temperature, spectral changes were observed that are consistent with ligand binding in axial position trans to the NO (the proximal site) and formation of the six-coordinate adducts. Analogous behavior was observed in solution. In both media, the six-coordinate adducts are stable only at low temperature and dissociate to the 5-coordinate nitrosyl complexes upon warming. The NO stretching frequencies of the six-coordinate thioether and ether complexes were recorded and binding constants for the weak bonding of proximal THF and THT ligands were determined from the spectral changes. These parameters are compared with those obtained for the N-donor ligand pyrrolidine.Graphical abstract shows formation of six-coordinate iron porphyrin nitrosyl complex with S-, O-, and N-donors upon lowering the temperature. Changes in the FTIR spectra of solution contained the five-coordinate iron porphyrin nitrosyl complex upon addition of N-donor followed by cooling.Highlights► Formation of Fe(TTP)(NO)L (L = S-, O-, N-donors) at low T is shown by FTIR and UV-Vis. ► Keq and thermodynamic parameters of reaction were determined for THT, THF and Pyrr. ► DFT computations of the S- and N- donor ligands binding to the heme nitrosyl model.
Co-reporter:R. Dale Rimmer, Agustin E. Pierri, Peter C. Ford
Coordination Chemistry Reviews 2012 Volume 256(15–16) pp:1509-1519
Publication Date(Web):August 2012
DOI:10.1016/j.ccr.2011.12.009
The potential therapeutic effects of carbon monoxide in several disease states, and new methodologies for CO delivery to specific targets is a topic of growing interest in mammalian physiology. Here we provide an overview of recent efforts in this laboratory regarding several types of carbonyl complexes of the group 6 metal and of those in other laboratories that are focused on developing photochemical techniques for such CO delivery. Photo-activated carbon monoxide releasing moieties (photoCORMs) based on manganese, iron and group 6 metals are described.Highlights► We describe desired properties for photochemical CO delivery to biological targets. ► We also describe analytical challenges encountered in developing photoCORMs. ► We outline previous investigations of photoCORMs published by us and others. ► We present a new class of photoCORMs where CO is labilized by visible light.
Co-reporter:Peter T. Burks ; Alexis D. Ostrowski ; Alexander A. Mikhailovsky ; Emory M. Chan ; Paul S. Wagenknecht
Journal of the American Chemical Society 2012 Volume 134(Issue 32) pp:13266-13275
Publication Date(Web):July 18, 2012
DOI:10.1021/ja300771w
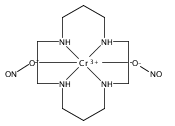
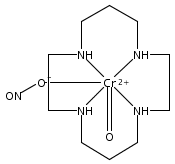
Reported are quantitative studies of the energy transfer from water-soluble CdSe/ZnS and CdSeS/ZnS core/shell quantum dots (QDs) to the Cr(III) complexes trans-Cr(N4)(X)2+ (N4 is a tetraazamacrocycle ligand, X– is CN–, Cl–, or ONO–) in aqueous solution. Variation of N4, of X–, and of the QD size and composition allows one to probe the relationship between the emission/absorption overlap integral parameter and the efficiency of the quenching of the QD photoluminescence (PL) by the chromium(III) complexes. Steady-state studies of the QD PL in the presence of different concentrations of trans-Cr(N4)(X)2+ indicate a clear correlation between quenching efficiency and the overlap integral largely consistent with the predicted behavior of a Förster resonance energy transfer (FRET)-type mechanism. PL lifetimes show analogous correlations, and these results demonstrate that spectral overlap is an important consideration when designing supramolecular systems that incorporate QDs as photosensitizers. In the latter context, we extend earlier studies demonstrating that the water-soluble CdSe/ZnS and CdSeS/ZnS QDs photosensitize nitric oxide release from the trans-Cr(cyclam)(ONO)2+ cation (cyclam = 1,4,8,11-tetraazacyclotetradecane) and report the efficiency (quantum yield) for this process. An improved synthesis of ternary CdSeS core/shell QDs is also described.
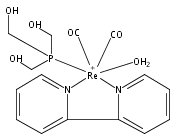
Co-reporter:Agustin E. Pierri ; Alessia Pallaoro ; Guang Wu
Journal of the American Chemical Society 2012 Volume 134(Issue 44) pp:18197-18200
Publication Date(Web):October 18, 2012
DOI:10.1021/ja3084434
The water-soluble rhenium(I) complex fac-[Re(bpy)(CO)3(thp)]+ (1) [CF3SO3– salt; bpy = 2,2′-bipyridine, thp = tris(hydroxymethyl)phosphine] is both strongly luminescent and photoactive toward carbon monoxide release. It is stable in aerated aqueous media, is incorporated into cells from the human prostatic carcinoma cell line PPC-1, and shows no apparent cytotoxicity. Furthermore, the solvated Re(I) photoproduct of CO release (2) is also luminescent, a feature that allows one to track the transformation of 1 to 2 inside such cells using confocal fluorescence microscopy. In this context, 1 is a very promising candidate as a photoactivated CO releasing moiety (photoCORM) with potential therapeutic applications.
Co-reporter:Tigran S. Kurtikyan, Vardan A. Hayrapetyan, Garik G. Martirosyan, Robert K. Ghazaryan, Alexei V. Iretskii, Hailiang Zhao, Kristine Pierloot and Peter C. Ford
Chemical Communications 2012 vol. 48(Issue 99) pp:12088-12090
Publication Date(Web):02 Nov 2012
DOI:10.1039/C2CC37337H
Reaction of NO with amorphous Mn(TPP) layers gives two Mn(TPP)(NO) isomers with linear and bent Mn–N–O geometries that reversibly interconvert with changes in temperature. DFT computations predict that the linear complex is the singlet ground state while the bent structure is a triplet state.
Co-reporter:Peter T. Burks and Peter C. Ford
Dalton Transactions 2012 vol. 41(Issue 42) pp:13030-13042
Publication Date(Web):17 Apr 2012
DOI:10.1039/C2DT30465A
Semiconductor quantum dots (QDs) are attractive for potential use as photosensitizers for a variety of applications. These nanomaterials have very high absorption cross-sections and often display strong photoluminescence (PL). Furthermore, QD absorption and emission spectra can be tuned simply by varying their size, and QD surfaces can be modified to access multiple sites for attaching potential acceptors as well as other functionalities. Here we provide an overview of recent studies concerned with the photosensitization of transition metal centers and other acceptors. Particular focus is directed towards potential therapeutic applications and to our own interest in the delivery of small molecule bioregulators to physiological targets. Studies that have addressed factors that control likely energy and charge transfer processes between QD donors and acceptor molecules are also discussed. Understanding the mechanisms of these photosensitization processes can provide design guidelines for successful applications.
Co-reporter:Julie L. Heinecke, Jun Yi, Jose Clayston Melo Pereira, George B. Richter-Addo, Peter C. Ford
Journal of Inorganic Biochemistry 2012 Volume 107(Issue 1) pp:47-53
Publication Date(Web):February 2012
DOI:10.1016/j.jinorgbio.2011.10.006
Nitrite reduction to nitric oxide by heme proteins is drawing increasing attention as a protective mechanism to hypoxic injury in mammalian physiology. Here we probe the nitrite reductase (NiR) activities of manganese(II)- and cobalt(II)-substituted myoglobins, and compare with data obtained previously for the iron(II) analog wt MbII. Both MnIIMb and CoIIMb displayed NiR activity, and it was shown that the kinetics are first order each in [protein], [nitrite], and [H+], as previously determined for the FeII analog wt MbII. The second order rate constants (k2) at pH 7.4 and T = 25 °C, were 0.0066 and 0.015 M− 1 s− 1 for CoIIMb and MnIIMb, respectively, both orders of magnitude slower than the k2 (6 M− 1 s− 1) for wt MbII. The final reaction products for MnIIMb consisted of a mixture of the nitrosyl MnIIMb(NO) and MnIIIMb, similar to the products from the analogous NiR reaction by wt Mb. In contrast, the products of NiR by CoIIMb were found to be the nitrito complex CoIIIMb(ONO−) plus roughly an equivalent of free NO. The differences can be attributed in part to the stronger coordination of inorganic nitrite to CoIIIMb as reflected in the respective MIIIMb(ONO−) formation constants Knitrite: 2100 M− 1 (CoIII) and <~0.4 M− 1 (MnIII). We also report the formation constants (3.7 and 30 M− 1, respectively) for the nitrite complexes of the mutant metmyoglobins H64V MbIII(NO2−) and H64V/V67R MbIII(ONO−) and a Knitrite revised value (120 M− 1) for the nitrite complex of wt metMb. The respective Knitrite values for the three ferric proteins emphasize the importance of a H-bonding residue, such as His64 in the MbIII distal pocket or the Arg67 in H64V/V67R MbIII, in stabilizing nitrite coordination. Notably, the NiR activities of the corresponding ferrous Mbs follow a similar sequence suggesting that nitrite binding to these centers are analogously affected by the H-bonding residues.Nitrite reduction to NO by wt Mb(II) and its metal-substituted compounds follows the order wt Mb ≫ MnMb > CoMb.Highlights► The conversion of nitrite to NO by Mn- and Co-substituted myoglobins was probed. ► The Mb-nitrite formation constants follow the order Co(III)Mb > Fe(III)Mb ≫ Mn(III)Mb. ► Both Mn(II)Mb and Co(II)Mb display nitrite reductase (NiR) reactivity. ► The order of NiR reactivity follows the order wt Mb(II) ≫ Mn(II)Mb > Co(II)Mb.
Co-reporter:Johannes R. Dethlefsen, Alexander A. Mikhailovsky, Peter T. Burks, Anders Døssing, and Peter C. Ford
The Journal of Physical Chemistry C 2012 Volume 116(Issue 44) pp:23713-23720
Publication Date(Web):October 29, 2012
DOI:10.1021/jp306915p
Lanthanide-modified CdSe quantum dots (CdSe(Ln) QDs) have been prepared by heating a solution of Cd(oleate)2, SeO2, and Ln(bipy)(S2CNEt2)3 (bipy = 2,2′-bipyridine) to 180–190 °C for 10–15 min. The elemental compositions of the resulting CdSe(Ln) cores and CdSe(Ln)/ZnS core/shell QDs show this route to be highly reproducible. The optical absorption spectra of these composite materials are similar to those of the unmodified nanocrystals, but the QD-centered band edge photoluminescence (PL) is partially quenched. The time-gated emission and excitation spectra of the CdSe(Ln) cores display sensitized lanthanide-centered PL upon higher energy excitation of the nanocrystal host but not upon excitation at the lowest energy QD absorption band. Growth of the ZnS shell led to the depletion of about 60% of the lanthanide ions present together with depletion of nearly all of the lanthanide-centered PL. On these bases, we conclude that the lanthanide-centered PL from the CdSe(Ln) cores originates with Ln3+-related trap states associated with the QD surface.
Co-reporter:Setsuko Kudo, James L. Bourassa, Susan E. Boggs, Yuhkou Sato, Peter C. Ford
Analytical Biochemistry 2012 420(2) pp: 197
Publication Date(Web):15 January 2012
DOI:10.1016/j.ab.2011.08.001
Co-reporter:Paul S. Wagenknecht, Peter C. Ford
Coordination Chemistry Reviews 2011 Volume 255(5–6) pp:591-616
Publication Date(Web):March 2011
DOI:10.1016/j.ccr.2010.11.016
Transition metal complexes are vital components in a wide range of photooptical applications; these range from targeted drug delivery to devices for the conversion of solar energy to electrical and/or stored chemical energy. Metal centered (MC) ligand field excited states play important roles in the photophysics of those complexes having partially filled d-orbitals. This review offers a broad perspective on key investigations that have characterized the chemistry and physics of MC excited states in d3 and d6 transition metal complexes. It will also illustrate the impact of these excited states on various photooptical applications and highlight efforts to understand, control, and tune these MC excited states in the context of such applications.Research highlights▶ The chemistry and physics of metal centered excited states in d3 and d6 TM complexes. ▶ Efforts to control and tune metal centered ligand field excited states. ▶ Metal centered excited states in targeted drug delivery schemes. ▶ Metal centered excited states involved in organic light emitting diodes. ▶ Metal centered excited states in the conversion of solar to stored chemical energy.
Co-reporter:Theodore D. Matson ; Katalin Barta ; Alexei V. Iretskii
Journal of the American Chemical Society 2011 Volume 133(Issue 35) pp:14090-14097
Publication Date(Web):August 1, 2011
DOI:10.1021/ja205436c
Efficient methodologies for converting biomass solids to liquid fuels have the potential to reduce dependence on imported petroleum while easing the atmospheric carbon dioxide burden. Here, we report quantitative catalytic conversions of wood and cellulosic solids to liquid and gaseous products in a single stage reactor operating at 300–320 °C and 160–220 bar. Little or no char is formed during this process. The reaction medium is supercritical methanol (sc-MeOH) and the catalyst, a copper-doped porous metal oxide, is composed of earth-abundant materials. The major liquid product is a mixture of C2–C6 aliphatic alcohols and methylated derivatives thereof that are, in principle, suitable for applications as liquid fuels.
Co-reporter:Alexis D. Ostrowski ; Ryan O. Absalonson ; Malcolm A. De Leo ; Guang Wu ; James G. Pavlovich ; Janet Adamson ; Bilal Azhar ; Alexei V. Iretskii ; Ian L. Megson
Inorganic Chemistry 2011 Volume 50(Issue 10) pp:4453-4462
Publication Date(Web):April 8, 2011
DOI:10.1021/ic200094x
Experimental and density functional theory (DFT) studies are described that are focused on outlining the reactivity of the known photochemical nitric oxide precursor trans-Cr(cyclam)(ONO)2+ (“CrONO”, cyclam = 1,4,8,11-tetrazacycltetradecane). Studies in both aerated and deaerated aqueous media are described as are the roles of both the oxidant O2 and a reductant such as glutathione in trapping the apparent Cr(IV) photoreaction intermediate trans-Cr(cyclam)(O)(ONO)+. Also reported and characterized structurally is the Cr(V) product of long-term photolysis in the absence of reducing agents, the trans-dioxo species [trans-Cr(cyclam)(O)2](ClO4). Photosensitization experiments indicate that at least a significant fraction of the reaction occurs from the lowest energy doublet excited state(s). Lastly, cell culture experiments demonstrate that CrONO has little or no acute toxicity either before or after photolysis.
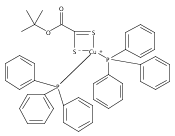
Co-reporter:David F. Zigler, Elisa Tordin, Guang Wu, Alexei Iretskii, Elena Cariati, Peter C. Ford
Inorganica Chimica Acta 2011 Volume 374(Issue 1) pp:261-268
Publication Date(Web):1 August 2011
DOI:10.1016/j.ica.2011.02.037
Described are the syntheses and structures of a phosphonium salt of the anionic ligand O-t-butyl-1,1-dithiooxalate, [PPh3Bz][i-dtotBu] ([PPh3Bz][1]), and of two Cu(I) complexes of this anion, Cu(PPh3)2(η2-i-dtotBu) (2) and Cu(dmp)(PPh3)(η1-i-dtotBu) (3, dmp = 2,9-dimethyl-1,10-phenanthroline). In addition, it was found that the reaction of CuBr2 with i-dtotBu− gives a O-t-butyl-1-perthio-1-thiooxalato complex of copper(I), [BzPh3P][Cu(Br)(S-i-dtotBu)] ([BzPh3P][4]), where [S-i-dtotBu]− is a disulfide-containing anionic ligand. The electronic structure and absorption spectrum of this species were investigated by time dependent DFT methods.Graphical abstractDescribed are syntheses and structures of a phosphonium salt of the anionic ligand O-t-butyl-1,1-dithiooxalate and of Cu(I) complexes of this anion. The reaction of CuBr2 with i-dtotBu− gives a O-t-butyl-1-perthio-1-thiooxalato complex of copper(I), [BzPh3P][Cu(Br)(S-i-dtotBu)] where [S-i-dtotBu]− is a disulfide-containing anionic ligand.Highlights► Salt and Cu(I) complexes of O-t-butyl-1,1, dithiooxalate (i-dtotBu-) described. ► i-dtotBu- reacts with CuBr2 to give perthio-1-thiooxalato complex with short S-S bond.Crystal structures are reported for i-dtotBu- and each Cu(I) complex.
Co-reporter:Peter C. Ford
Coordination Chemistry Reviews 2010 Volume 254(3–4) pp:195-196
Publication Date(Web):February 2010
DOI:10.1016/j.ccr.2009.08.002
Co-reporter:Julie Heinecke
Journal of the American Chemical Society 2010 Volume 132(Issue 27) pp:9240-9243
Publication Date(Web):June 21, 2010
DOI:10.1021/ja102221e
Cysteine sulfenic acid CysS(O)H is shown to be formed for the reaction of cysteine (CysSH) with aqueous nitrite and the water-soluble ferriheme models FeIII(TPPS) (TPPS = meso-tetra(4-sulfonatophenyl)porphyrinato) or FeIII(TMPS) (TMPS = meso-tetra(sulfonatomesityl)porphyrinato) at pH 5.8 and 7.4. The other product is the respective ferrous nitrosyl complex FeII(Por)(NO) (Por = TPPS or TMPS). Analogous oxygen atom transfers (OAT) were seen when glutathione (GSH) was used as the substrate. The sulfenic acids, CysS(O)H and GS(O)H, are transient species since they react rapidly with excess thiol to give the respective disulfides, so their presence as reactive intermediates was demonstrated by trapping with dimedone and detecting the resulting adduct using LC/MS. Preliminary kinetics studies are consistent with rate-limiting OAT from a ferric nitro complex FeIII(Por)(NO2−) to CysSH, although this reaction is complicated by a competing dead-end equilibrium to form the thiolate complex (FeIII(TPPS)(CysS−).
Co-reporter:Julie Heinecke, Peter C. Ford
Coordination Chemistry Reviews 2010 Volume 254(3–4) pp:235-247
Publication Date(Web):February 2010
DOI:10.1016/j.ccr.2009.07.021
This review provides a summary of reaction mechanisms involving the interactions of nitrite ion with metal centers relevant to physiology. The majority of the systems that have been investigated are heme proteins and models, where nitrite reacts with the central metal ions to generate important iron-NOx intermediates and subsequent NOx products. We also discuss reactions with other potentially relevant systems. Nitrite is formed as a product of NO autoxidation in aqueous media and can be formed by the bacterial reduction of ingested nitrate as well. It is now generally accepted that under certain conditions nitrite, which is present in mammalian fluids and tissue at micromolar concentrations, can serve as a biological reserve of the bioregulatory agent nitric oxide. However, it is possible that nitrite serves other functions as well. The goal of this review is to evaluate the present state of understanding regarding these pathways and the delicate interplay between nitrite and the various NOx species of biological relevance.
Co-reporter:Katalin Barta, Theodore D. Matson, Makayla L. Fettig, Susannah L. Scott, Alexei V. Iretskii and Peter C. Ford
Green Chemistry 2010 vol. 12(Issue 9) pp:1640-1647
Publication Date(Web):10 Aug 2010
DOI:10.1039/C0GC00181C
A novel approach to disassembling biomass-derived lignin into processible units is described. This transformation is achieved in supercritical methanol, using a Cu-doped porous metal oxide as the catalyst, at a relatively mild temperature (300 °C). Hydrogen transfer from methanol to an organosolv lignin results in the complete hydrogenolysis of phenyl ether bonds, coupled with the hydrogenation of aromatic rings. The product is a complex mixture composed principally of monomeric substituted cyclohexyl derivatives with greatly reduced oxygen content and negligible aromatics. Notably, no char formation was observed. We also describe operational indices based on the 1H NMR spectra that facilitate holistic evaluation of the product distribution in this and other biomass transformations.
Co-reporter:Tigran S. Kurtikyan and Peter C. Ford
Chemical Communications 2010 vol. 46(Issue 45) pp:8570-8572
Publication Date(Web):21 Oct 2010
DOI:10.1039/C0CC02665D
The oxy-globin models Fe(Por)(NH3)(O2), prepared by sequential reactions of O2 (18O2) and NH3 with thin porous layers of FeII(Por), react with NO (15NO) at 80–100 K to form only the low-spin nitrato complexes Fe(Por)(NH3)(η1-ONO2), thus implying that peroxynitrite intermediates, if formed, must undergo very facile isomerization to the nitrato analog.
Co-reporter:Alexis D. Ostrowski ; Sherine J. Deakin ; Bilal Azhar ; Thomas W. Miller ; Nestor Franco ; Melisa M. Cherney ; Andrea J. Lee ; Judith N. Burstyn ; Jon M. Fukuto ; Ian L. Megson
Journal of Medicinal Chemistry 2010 Volume 53(Issue 2) pp:715-722
Publication Date(Web):December 1, 2009
DOI:10.1021/jm9013357
The chromium(III) nitrito complex trans-Cr(cyclam)(ONO)2+ (1) is a very promising photochemical precursor for nitric oxide delivery to physiological targets. Here, we demonstrate that visible wavelength excitation of 1 in solutions containing thiol reductants such as the biological antioxidant glutathione (GSH) leads to permanent reaction even under anaerobic conditions, resulting in high quantum yield NO release. The nitric oxide formed under such conditions is sufficient, even at μM concentrations of 1 and using a low-intensity light source, to activate the enzyme soluble guanylyl cyclase (sGC). We also demonstrate that photolysis of 1 in the nM concentration range with a portable blue LED leads to vasorelaxation of porcine coronary arterial rings, a process also attributed to the NO activation of sGC.
Co-reporter:Peter C. Ford
Inorganic Chemistry 2010 Volume 49(Issue 14) pp:6226-6239
Publication Date(Web):July 12, 2010
DOI:10.1021/ic902073z
Presented is an overview of the fundamental chemical properties of nitric oxide and nitrite ion in relation to the reactions with ferri- and ferroheme models and proteins potentially relevant to the roles of these species in mammalian biology.
Co-reporter:R. Dale Rimmer ; Heinrich Richter
Inorganic Chemistry 2010 Volume 49(Issue 3) pp:1180-1185
Publication Date(Web):December 29, 2009
DOI:10.1021/ic902147n
Interest in therapeutic applications of carbon monoxide release to physiological targets has led us to explore a photochemical strategy for such CO delivery. Here, we describe the photoactivated carbon monoxide releasing moiety (photoCORM), W(CO)5(TPPTS)3− (1), an air-stable, water-soluble tungsten(0) carbonyl complex of the trianionic ligand tris(sulphonatophenyl)phosphine. Near-UV photolysis of 1 in an aqueous buffer solution leads to the high quantum yield release of a single CO, the formation of which has been verified by three analytical methodologies. Furthermore, in aerated media, additional CO is slowly released from the W(CO)4(H2O)(TPPTS)3− photoproduct owing to autoxidation of the tungsten center. Thus, 1 serves as a carbon monoxide releasing moiety both in the primary photochemical reaction and in the secondary reactions of the initially formed photoproduct. The three methodologies for quantifying CO release under these physiologically relevant conditions are also described.
Co-reporter:Johannes W. Dethlefsen, Erik D. Hedegård, R. Dale Rimmer, Peter C. Ford and Anders Døssing
Inorganic Chemistry 2009 Volume 48(Issue 1) pp:231-238
Publication Date(Web):November 25, 2008
DOI:10.1021/ic8016936
Photolysis of the thionitrosyl complex Cr(CH3CN)5(NS)2+ (1) in acetonitrile solution leads to the dissociation of nitrogen monosulfide (NS). In deaerated solution, this reaction is reversible, and flash photolysis studies demonstrate that NS reacts with Cr(CH3CN)62+ according to the rate law d[1]/dt = kon[Cr(CH3CN)62+][NS] (kon = 2.3 × 108 M−1 s−1 at 298 K). The photolysis of 1 in deaerated acetonitrile with added Fe(S2CNEt2)2 leads to the transfer of NS and the formation of a species concluded to be Fe(S2CNEt2)2(NS) based on its electron paramagnetic resonance spectrum. Analogous photolysis of 1 in the presence of added NO leads to clean formation of the nitrosyl complex Cr(CH3CN)5(NO)2+ (2) presumably by NO capture of the photoproduct Cr(CH3CN)62+ (3). When 1 was photolyzed in aerated acetonitrile solution, the reactive species 3 was trapped, thus leading to net photochemical transformations with excitation-wavelength-dependent quantum yields of 0.3−1.0 mol/Einstein. Mass spectroscopic studies of the product solutions demonstrate the formation of S8, presumably from the decomposition of NS. The quantitative photochemical behaviors of 1 and the nitrosyl analog 2 are compared.
Co-reporter:Tigran S. Kurtikyan ; Astghik A. Hovhannisyan ; Alexei V. Iretskii
Inorganic Chemistry 2009 Volume 48(Issue 23) pp:11236-11241
Publication Date(Web):November 3, 2009
DOI:10.1021/ic901722g
Spectroscopic studies demonstrate that the 5-coordinate O-nitrito complexes Fe(Por)(η1-ONO) (Por - meso-tetraphenyl- or meso-tetra-p-tolyl-porphyrinato dianions) react with the thioethers (R2S) dimethylsulfide and tetrahydrothiophene to give the 6-coordinate N-nitrito complexes Fe(Por)(R2S)(NO2). These reactions were conducted in low-temperature porous layered solids formed in a cryostat; however, with excess R2S in the atmosphere, the same species are moderately stable at room temperature. Six-coordinate O-nitrito isomers were not observed with the R2S proximal ligands, even though DFT calculations for the Fe(P)(DMS)(η1-ONO) and Fe(P)(DMS)(NO2) models (P = porphinato dianion, DMS = dimethyl sulfide) show the latter to be only modestly lower energy (∼8 kJ/mol) than the former. Leaving this system at room temperature in the presence of excess R2S leads eventually to the appearance in the FTIR spectra of the ν(NO) band characteristic of the ferrous nitrosyl Fe(Por)(NO). Concomitantly, the mass spectrum of the gas phase demonstrated the molecular peaks of the sulfoxides R2SO, indicating oxygen atom transfer reactivity for the ferric porphryinato complexes of nitrite.
Co-reporter:Alexis D. Ostrowski and Peter C. Ford
Dalton Transactions 2009 (Issue 48) pp:10660-10669
Publication Date(Web):27 Aug 2009
DOI:10.1039/B912898K
The bioregulatory molecule NO plays key roles in cancer biology and has been implicated in both tumor growth and suppression. Furthermore, it is a γ-radiation sensitizer that may enhance selective killing of neoplastic tissues. For these reasons, there is considerable interest in developing methods for NO delivery to specific physiological targets. In this Perspective, we describe ongoing investigations focused on photochemical methodologies to deliver therapeutic doses of NO to such targets utilizing transition metal complexes that are nitric oxide precursors. The photochemical strategy has the advantages that it allows for precise control of the timing, location, and dosage for the targeted delivery of a bioactive agent.
Co-reporter:Birgit Birkmann, Bridget T. Owens, Susmita Bandyopadhyay, Guang Wu, Peter C. Ford
Journal of Inorganic Biochemistry 2009 Volume 103(Issue 2) pp:237-242
Publication Date(Web):February 2009
DOI:10.1016/j.jinorgbio.2008.10.012
Reaction of silver nitrite with Ru(salen)(PPh3)(Cl) (salen2− = N,N′-ethylenebis(salicylideneiminato dianion) using [NEt4]OH as a phase transfer agent led to the formation of a stable, N-coordinated nitrite ion complex (RuIII–NO2) that was characterized by single crystal X-ray diffraction. This reaction also resulted in nitration of the two phenolic rings of the salen ligand, which was unexpected given the basic conditions. With sodium nitrite, the Ru(III)-nitro complex was formed, but no ring nitration was observed. Thus, in a non-acidic medium, the combination of nitrite with redox active metal centers may provide a route to the nitration of phenolic derivatives. Possible mechanisms of this unusual reaction having potential relevance to aromatic nitrations in biological systems are discussed. This is also the first report of the crystal structure of a Ru(III)-nitro complex, a species that has been suggested to be unstable and prone to rapid disproportionation.
Co-reporter:GeraldS. Macala;TheodoreD. Matson;CharlesL. Johnson;RobertS. Lewis;AlexeiV. Iretskii ;PeterC. Ford
ChemSusChem 2009 Volume 2( Issue 3) pp:215-217
Publication Date(Web):
DOI:10.1002/cssc.200900033
Co-reporter:Peter C. Ford
Accounts of Chemical Research 2008 Volume 41(Issue 2) pp:190
Publication Date(Web):January 9, 2008
DOI:10.1021/ar700128y
In order to deliver a bioactive agent to a physiological location, it is important to be able to regulate precisely the location and the dosage. Such exquisite control can easily be envisioned for a photochemical drug that is active toward release of the desired bioactive agent upon irradiation of a specific tissue site. These materials should be thermally stable but reactive under excitation at visible (vis) or near-infrared (NIR) wavelengths where tissue transmission is optimal. Two photon excitation (TPE) is of special interest, since the use of focused laser pulses to activate release could provide 3D spatial control in therapeutic applications. This Account describes the preparation and photochemistry of a series of transition metal complexes designed to release the simple bioregulatory compound nitric oxide upon vis or NIR excitation. In order to enhance the light gathering capability of such compounds, we have attached chromophores with high single- or two-photon absorption cross sections to several photochemical NO precursors. For example, the iron nitrosyl clusters Fe2(μ-SR)2(NO)4 (Roussin’s red esters) have been prepared with various chromophores as pendant groups, an example being the protoporphyrin XI derivative illustrated here. Direct excitation into the vis absorbing Q bands of the porphyrin leads to enhanced rates of NO generation from the Fe/S/NO cluster owing to the larger rate of light absorption by that antenna. Furthermore, femtosecond pulsed laser NIR excitation of the same compound at 810 nm (a spectral region where no absorption bands are apparent) leads to weak emission at ~630 nm and generation of NO, both effects providing evidence of a TPE mechanism. Roussin’s red esters with other chromophores described here are even more effective for TPE-stimulated NO release. Another photochemical NO precursor discussed is the Cr(III) complex trans-Cr(L)(ONO)2+ where L is a cyclic tetraamine such as cyclam. When L includes a chromophore tethered to the ligand backbone, excitation of that functionality results in energy transfer to the spin-forbidden ligand field double states and light-stimulated release of NO. We are working to develop systems where L is attached to a semiconductor nanoparticle as the antenna. In this context, we have shown that electrostatic assemblies are formed between the anionic surface of water-soluble CdSe/ZnS core/shell quantum dots (QDs) and Cr(L)(ONO)2+ cations via an ion-pairing mechanism. Photoexcition of such modified QDs leads to markedly enhanced NO generation and suggests promising applications of such nanomaterials as photochemical drugs.
Co-reporter:Tigran S. Kurtikyan, Peter C. Ford
Coordination Chemistry Reviews 2008 Volume 252(12–14) pp:1486-1496
Publication Date(Web):July 2008
DOI:10.1016/j.ccr.2007.10.012
Porous layered solids can be prepared by the vacuum sublimation of the ferrous porphyrin complexes Fe(Por) onto a CaF2 or KBr substrate in a cryostat. The facile reactions of these heme models with volatile reactants can be studied in detail using FTIR and optical spectroscopy. The power of this technique draws from the ability to investigate such processes under carefully controlled and tunable conditions, especially temperature. Furthermore, the solvent-free medium gives relatively sharp bands in the FTIR spectra that, combined with isotopic labeling experiments, provide structural information regarding otherwise elusive intermediates and insight into key mechanistic steps. Reviewed here are investigations of the reactions between various Fe(Por) and the nitrogen oxides NO and NO2 to give (initially) such species as Fe(Por)(NO), Fe(Por)(η1-ONO) and Fe(Por)(η2-O2NO). Also described are subsequent transformations of these upon exposure to NO, NO2 and various Lewis bases. These simple models provide fundamental information relevant to the reactions of heme proteins with the biologically important nitrogen oxides.
Co-reporter:Enrique Lozano Diz, Peter C. Ford
Inorganica Chimica Acta 2008 Volume 361(Issue 11) pp:3084-3088
Publication Date(Web):27 July 2008
DOI:10.1016/j.ica.2008.01.035
Kinetics studies of the behavior of Mo(CO)5(CH) in the presence of radical initiators were conducted in cyclohexane (CH) solution by laser flash photolysis with time-resolved infrared detection. Activation parameters were determined for reactions of Mo(CO)5(CH) with toluene and with photochemical radical generator dibenzylketone (DBK) in the presence of excess CO.Flash photolysis of Mo(CO)6 in cyclohexane (CH) solution leads to the reversible formation of the pentacarbonyl complex Mo(CO)5(CH) (I). I reacts with toluene or dibenzylketone to give Mo(CO)5(L) with rates and activation parameters indicating a dissociative interchange mechanism. Simultaneous generation of benzyl radicals had little effect on these pathways.
Co-reporter:Gerald S. Macala;Andrew W. Robertson;Charles L. Johnson
Catalysis Letters 2008 Volume 122( Issue 3-4) pp:205-209
Publication Date(Web):2008 May
DOI:10.1007/s10562-008-9480-y
Described are new solid base catalysts for transesterification of seed oil triglycerides to fatty acid methyl esters, a key step in biodiesel production. These were prepared by substituting Fe3+ ions substitute for a fraction of the Al3+ ions in the Mg/Al layered double hydroxide lattices of hydrotalcites (HTC) and calcining to give porous metal oxides (PMOs). These iron-doped PMOs are much stronger bases than those derived from undoped or Ga3+ doped HTCs and are effective catalysts for the methanol transesterification of triacetin (glycerol triacetate) and of soybean oil.
Co-reporter:Jon Marhenke, Steven M. Massick, Peter C. Ford
Inorganica Chimica Acta 2007 Volume 360(Issue 3) pp:825-836
Publication Date(Web):15 February 2007
DOI:10.1016/j.ica.2006.05.023
Flash photolysis with time-resolved infrared (TRIR) spectroscopy was used to elucidate the photochemical reactivity of the hydroformylation catalyst precursor Co2(CO)6(PMePh2)2. Depending on reaction conditions, the net products of photolysis varied significantly. A model is presented that accounts for the net reactivity with two initial photoproducts, the 17-electron species Co(CO)3(PMePh2) and the coordinatively unsaturated dimer Co2(CO)5(PMePh2)2. No evidence was found for photochemical formation of Co2(CO)6(PMePh2). Time-resolved spectroscopic studies allowed for the direct observation of transient species and for kinetics studies of certain reactions; for example, the reactions of Co(CO)3PMePh2 with CO and with PMePh2 gave the respective rate constants 1.5 × 105 and 1.2 × 107 M−1 s−1, while the analogous reactions with Co2(CO)5(PMePh2)2 gave the rate constants of 2.6 × 106 M−1 s−1 and 3.9 × 107 M−1 s−1.Flash photolysis with time-resolved infrared spectroscopy was used to elucidate the reactivity of radicals and coordinatively unsaturated intermediates generated from Co2(CO)6(PMePh2)2.
Co-reporter:Roberto Santana da Silva, Mario Sergio P. Marchesi, Antonio Claudio Tedesco, Alexander Mikhailovsky and Peter C. Ford
Photochemical & Photobiological Sciences 2007 vol. 6(Issue 5) pp:515-518
Publication Date(Web):20 Mar 2007
DOI:10.1039/B617350K
Metal-to-ligand charge transfer photolysis of the ruthenium(II) pyrazine complex Ru(NH3)5pz2+ (I) in pH 7.4 oxygenated phosphate buffer solution generates the Ru(III) analog Ru(NH3)5pz3+ plus the reactive oxygen species singlet oxygen and superoxide. Based on the very short MLCT lifetime (re-measured as ∼250 ps in D2O) of I* and the quantum yield for singlet oxygen formation (0.01 for aerated D2O) the rate constant for oxygen quenching of I* was calculated to be ∼(3 ± 1) × 1010 M−1 s−1.
Co-reporter:Tigran S. Kurtikyan Dr. Dr.
Angewandte Chemie 2006 Volume 118(Issue 3) pp:
Publication Date(Web):2 DEC 2005
DOI:10.1002/ange.200502409
ONO oder O2N: Die schwierig nachzuweisenden fünffach koordinierten Nitritokomplexe [FeIII(por)(ONO)] (por=meso-Tetraphenyl- oder meso-Tetra-p-tolylporphyrinat-Dianion) bildeten sich bei der Wechselwirkung von Niederdruck-NO2 mit sublimierten [FeII(por)]-Schichten. Spektroskopische Daten bestätigen, dass der NO2−-Ligand bevorzugt über O bindet, dass er aber die N-gebundene Konfiguration annimmt, wenn die zweite axiale Position besetzt ist (R=Ph, p-Tolyl).
Co-reporter:Tigran S. Kurtikyan,Peter C. Ford
Angewandte Chemie International Edition 2006 45(3) pp:492-496
Publication Date(Web):
DOI:10.1002/anie.200502409
Co-reporter:Garik G. Martirosyan, Arsen S. Azizyan, Tigran S. Kurtikyan and Peter C. Ford
Chemical Communications 2004 (Issue 13) pp:1488-1489
Publication Date(Web):24 May 2004
DOI:10.1039/B404491F
Reaction of NO gas with sublimed layers of the MnIITPP (TPP =
meso-tetraphenylporphyrinato2−) at low temperature leads to nitric oxide disproportionation. UV-Vis and FTIR spectroscopy with isotopically substituted nitrogen oxides revealed formation of the unstable species identified as trans-MnIII(TPP)(NO)(ONO).
Co-reporter:Xiao-Ping Zhou, Aysen Yilmaz, Gurkan A. Yilmaz, Ivan M. Lorkovic, Leroy E. Laverman, Michael Weiss, Jeffrey H. Sherman, Eric W. McFarland, Galen D. Stucky and Peter C. Ford
Chemical Communications 2003 (Issue 18) pp:2294-2295
Publication Date(Web):13 Aug 2003
DOI:10.1039/B307235E
The partial oxidation of alkanes via bromination followed by the reaction with solid metal oxide mixtures (MO) is shown to give an array of products that can be tuned by varying the MO and the reaction conditions.
Co-reporter:Tigran S. Kurtikyan, Garik G. Martirosyan, Manya E. Hakobyan and Peter C. Ford
Chemical Communications 2003 (Issue 14) pp:1706-1707
Publication Date(Web):13 Jun 2003
DOI:10.1039/B302061D
Reaction of NO gas with low temperature films of the η2-nitrato model heme FeIII(TPP)(O2NO)
(TPP =
meso-tetraphenylporphyrinato2−) leads to formation of the previously unknown η1-nitrato nitrosyl species FeIII(TPP)(ONO2)(NO) as characterized by IR and optical spectroscopy with isotopically substituted nitrogen oxides.
Co-reporter:Peter C. Ford
Nitric Oxide (1 November 2013) Volume 34() pp:56-64
Publication Date(Web):1 November 2013
DOI:10.1016/j.niox.2013.02.001
There remains considerable interest in developing methods for the targeted delivery of nitric oxide and other small molecule bioregulators such as carbon monoxide to physiological targets. One such strategy is to use a “caged” NO that is “uncaged” by excitation with light. Such photochemical methods convey certain key advantages such as the ability to control the timing, location and dosage of delivery, but also have some important disadvantages, such as the relatively poor penetration of the ultraviolet and visible wavelengths often necessary for the uncaging process. Presented here is an overview of ongoing studies in the author’s laboratory exploring new photochemical NO precursors including those with nanomaterial antennas designed to enhance the effectiveness of these precursors with longer excitation wavelengths.Highlights► Several photochemical NO precursors (“caged” NO) are reviewed. ► Antenna chromophores enhance rates of NO uncaging. ► Two photon absorption with tissue penetrating NIR light can induce NO release. ► Nanocarriers incorporating NIR-to-visible upconversion are effective for NO uncaging.
Co-reporter:Elizabeth S. Levy, Demosthenes P. Morales, John V. Garcia, Norbert O. Reich and Peter C. Ford
Chemical Communications 2015 - vol. 51(Issue 100) pp:NaN17695-17695
Publication Date(Web):2015/10/16
DOI:10.1039/C5CC07989F
We demonstrate modulation of nitric oxide release in solution and in human prostate cancer cells from a thiol functionalized cupferron (TCF) absorbed on hollow gold nanoshells (HGNs) using near-infrared (NIR) light. NO release from the TCF–HGN conjugates occurs through localized surface heating due to NIR excitation of the surface plasmon. Specific HGN targeting is achieved through cell surface directed peptides, and excitation with tissue penetrating NIR light provides unprecedented spatio-temporal control of NO delivery to biological targets.
Co-reporter:Tigran S. Kurtikyan and Peter C. Ford
Chemical Communications 2010 - vol. 46(Issue 45) pp:NaN8572-8572
Publication Date(Web):2010/10/21
DOI:10.1039/C0CC02665D
The oxy-globin models Fe(Por)(NH3)(O2), prepared by sequential reactions of O2 (18O2) and NH3 with thin porous layers of FeII(Por), react with NO (15NO) at 80–100 K to form only the low-spin nitrato complexes Fe(Por)(NH3)(η1-ONO2), thus implying that peroxynitrite intermediates, if formed, must undergo very facile isomerization to the nitrato analog.
Co-reporter:Alexis D. Ostrowski and Peter C. Ford
Dalton Transactions 2009(Issue 48) pp:NaN10669-10669
Publication Date(Web):2009/08/27
DOI:10.1039/B912898K
The bioregulatory molecule NO plays key roles in cancer biology and has been implicated in both tumor growth and suppression. Furthermore, it is a γ-radiation sensitizer that may enhance selective killing of neoplastic tissues. For these reasons, there is considerable interest in developing methods for NO delivery to specific physiological targets. In this Perspective, we describe ongoing investigations focused on photochemical methodologies to deliver therapeutic doses of NO to such targets utilizing transition metal complexes that are nitric oxide precursors. The photochemical strategy has the advantages that it allows for precise control of the timing, location, and dosage for the targeted delivery of a bioactive agent.
Co-reporter:Peter T. Burks and Peter C. Ford
Dalton Transactions 2012 - vol. 41(Issue 42) pp:NaN13042-13042
Publication Date(Web):2012/04/17
DOI:10.1039/C2DT30465A
Semiconductor quantum dots (QDs) are attractive for potential use as photosensitizers for a variety of applications. These nanomaterials have very high absorption cross-sections and often display strong photoluminescence (PL). Furthermore, QD absorption and emission spectra can be tuned simply by varying their size, and QD surfaces can be modified to access multiple sites for attaching potential acceptors as well as other functionalities. Here we provide an overview of recent studies concerned with the photosensitization of transition metal centers and other acceptors. Particular focus is directed towards potential therapeutic applications and to our own interest in the delivery of small molecule bioregulators to physiological targets. Studies that have addressed factors that control likely energy and charge transfer processes between QD donors and acceptor molecules are also discussed. Understanding the mechanisms of these photosensitization processes can provide design guidelines for successful applications.
Co-reporter:Tigran S. Kurtikyan, Vardan A. Hayrapetyan, Garik G. Martirosyan, Robert K. Ghazaryan, Alexei V. Iretskii, Hailiang Zhao, Kristine Pierloot and Peter C. Ford
Chemical Communications 2012 - vol. 48(Issue 99) pp:NaN12090-12090
Publication Date(Web):2012/11/02
DOI:10.1039/C2CC37337H
Reaction of NO with amorphous Mn(TPP) layers gives two Mn(TPP)(NO) isomers with linear and bent Mn–N–O geometries that reversibly interconvert with changes in temperature. DFT computations predict that the linear complex is the singlet ground state while the bent structure is a triplet state.
Co-reporter:Agustin E. Pierri, Po-Ju Huang, John V. Garcia, James G. Stanfill, Megan Chui, Guang Wu, Nanfeng Zheng and Peter C. Ford
Chemical Communications 2015 - vol. 51(Issue 11) pp:NaN2075-2075
Publication Date(Web):2014/12/16
DOI:10.1039/C4CC06766E
A water-soluble nanocarrier for a photo-activated CO releasing moiety (photoCORM) that can be triggered with NIR excitation is described. This has an upconversion nanoparticle core encapsulated by an amphiphilic polymer imparting both water solubility and a hydrophobic interior containing the photoCORM trans-Mn(bpy)(PPh3)2(CO)2. Such an ensemble offers a unique strategy for CO delivery to biological targets.
Co-reporter:Christopher M. Bernt, Giovanni Bottari, Jacob A. Barrett, Susannah L. Scott, Katalin Barta and Peter C. Ford
Catalysis Science & Technology (2011-Present) 2016 - vol. 6(Issue 9) pp:NaN2994-2994
Publication Date(Web):2015/11/27
DOI:10.1039/C5CY01555C
Copper-doped porous metal oxides catalyze the one-pot disassembly of biomass-derived lignin via C–O bond hydrogenolysis and hydrodeoxygenation in supercritical methanol. This catalytic system cleanly converts lignin as well as lignocellulose composites, such as sawdust, to organic liquids with little or no formation of intractable tars or chars. However, this catalyst based on Earth-abundant components also catalyzes less desirable aromatic ring hydrogenations and various methylations that contribute to the diversity of products. In this context, we undertook a quantitative experimental and computational evaluation of model reactions relevant to the reductive disassembly of lignin by this catalyst system in order to determine quantitatively the rates of desirable and less desirable chemical steps that define the overall product selectivities. Global fitting analysis methods were used to map the temporal evolution of key intermediates and products and to elucidate networks that provide guidelines regarding the eventual fates of reactive intermediates in this catalysis system. Phenolic compounds display multiple reaction pathways, but substrates such as benzene, toluene, and alkyl- and alkoxy-substituted aromatics are considerably more stable under these conditions. These results indicate that modifying this catalytic system in a way that controls and channels the reactivity of phenolic intermediates should improve selectivity toward producing valuable aromatic chemicals from biomass-derived lignin. To this end we demonstrate that the O-methylating agent dimethyl carbonate can intercept the phenol intermediate formed from hydrogenolysis of the model compound benzyl phenyl ether. Trapping the phenol as anisole thus gave much higher selectivity towards aromatic products.
Co-reporter:Anthony W. DeMartino, David F. Zigler, Jon M. Fukuto and Peter C. Ford
Chemical Society Reviews 2017 - vol. 46(Issue 1) pp:NaN39-39
Publication Date(Web):2016/09/27
DOI:10.1039/C6CS00585C
The overview presented here has the goal of examining whether carbon disulfide (CS2) may play a role as an endogenously generated bioregulator and/or has therapeutic value. The neuro- and reproductive system toxicity of CS2 has been documented from its long-term use in the viscose rayon industry. CS2 is also used in the production of dithiocarbamates (DTCs), which are potent fungicides and pesticides, thus raising concern that CS2 may be an environmental toxin. However, DTCs also have recognized medicinal use in the treatment of heavy metal poisonings as well as having potency for reducing inflammation. Three known small molecule bioregulators (SMBs) nitric oxide, carbon monoxide, and hydrogen sulfide were initially viewed as environmental toxins. Yet each is now recognized as having intricate, though not fully elucidated, biological functions at concentration regimes far lower than the toxic doses. The literature also implies that the mammalian chemical biology of CS2 has broader implications from inflammatory states to the gut microbiome. On these bases, we suggest that the very nature of CS2 poisoning may be related to interrupting or overwhelming relevant regulatory or signaling process(es), much like other SMBs.
.jpg)


![Ethanethioic acid, S-[(4-nitrophenyl)methyl] ester](http://img.cochemist.com/ccimg/170700/170640-75-2.png)
![Ethanethioic acid, S-[(4-nitrophenyl)methyl] ester](http://img.cochemist.com/ccimg/170700/170640-75-2_b.png)





![L-Ornithine,N5-[imino(nitroamino)methyl]-, methyl ester](http://img.cochemist.com/ccimg/51000/50903-99-6.png)
![L-Ornithine,N5-[imino(nitroamino)methyl]-, methyl ester](http://img.cochemist.com/ccimg/51000/50903-99-6_b.png)





















































































![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8_b.png)