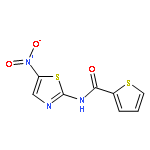
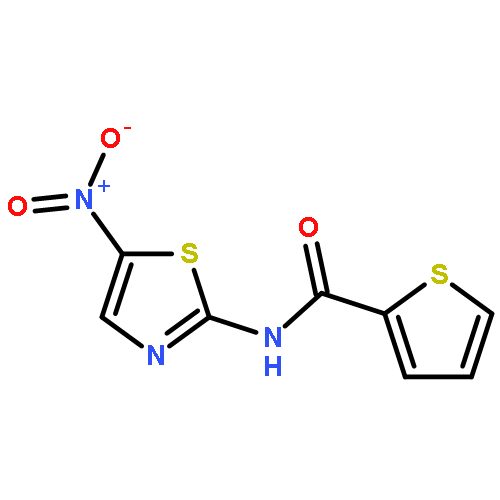
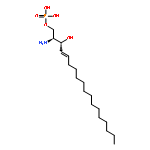
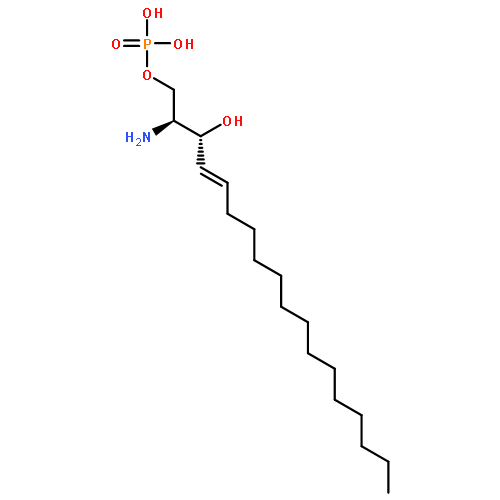
Co-reporter:Thomas P. Mathews ; Andrew J. Kennedy ; Yugesh Kharel ; Perry C. Kennedy ; Oana Nicoara ; Manjula Sunkara ; Andrew J. Morris ; Brian R. Wamhoff ; Kevin R. Lynch
Journal of Medicinal Chemistry 2010 Volume 53(Issue 7) pp:2766-2778
Publication Date(Web):March 5, 2010
DOI:10.1021/jm901860h
Sphingosine 1-phosphate (S1P), a potent phospholipid growth and trophic factor, is synthesized in vivo by two sphingosine kinases. Thus these kinases have been proposed as important drug targets for treatment of hyperproliferative diseases and inflammation. We report here a new class of amidine-based sphingosine analogues that are competitive inhibitors of sphingosine kinases exhibiting varying degrees of enzyme selectivity. These inhibitors display KI values in the submicromolar range for both sphingosine kinases and, in cultured vascular smooth muscle cells, decrease S1P levels and initiate growth arrest.
Co-reporter:Min Yang, Mahendra D. Chordia, Fengping Li, Tao Huang, Joel Linden, and Timothy L. Macdonald
Chemical Research in Toxicology 2010 Volume 23(Issue 11) pp:1691
Publication Date(Web):October 12, 2010
DOI:10.1021/tx1001496
Nimesulide, a widely used nonsteroidal anti-inflammatory drug (NSAID), has been associated with rare idiosyncratic hepatotoxicity. The chemical mechanisms underlying the liver injury remain unknown. We have undertaken the detailed study of the metabolic pathways of nimesulide in an effort to identify potential reactive metabolites. A previous report from this laboratory has demonstrated that one of the known nimesulide metabolites, termed reduced nimesulide (M1), is further bioactivated by human liver microsomes (HLMs) to form a reactive diiminoquinone species M2. The formation of M2 was confirmed indirectly by trapping with N-acetylcysteine (NAC). The aim of this study was to explore the fate of M1 in an inflammatory environment created by the recruitment of leukocytes. Leukocytes upon activation produce hydrogen peroxide (H2O2) and other myeloperoxidase (MPO) products, such as hypochlorous acid (HOCl), that are capable of metabolite oxidation. We demonstrate here that the reduced nimesulide, M1, undergoes a facile oxidation with activated neutrophils or with MPO in the presence of H2O2 or HOCl to produce a variety of reactive as well as stable metabolites. One major metabolite, M3, was also produced by HLM as determined by trapping with NAC. Other metabolites, for example, M6, M8, and M9, were unique to the myeloperoxidase, because of their mode of formation from activation of the amino group of reduced nimesulide. The structures of some of these reactive metabolites were proposed on the basis of liquid chromatography−tandem mass spectrometry analyses and established by their comparison with synthetic standards. Metabolite M6 is interesting because it provides clear evidence of amine activation and indicates the potential of the reactive intermediate of M1 to conjugate with protein nucleophiles. In summary, our results demonstrate that a known nimesulide metabolite could be bioactivated by MPO through a pathway distinct from HLM-mediated pathways and that the generation of reactive species by the MPO-mediated bioactivation pathway at the site of inflammation may contribute to the toxicity associated with nimesulide.
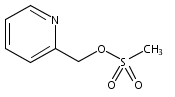
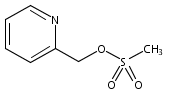
Co-reporter:James E. East, Andrew J. Kennedy, Jose L. Tomsig, Alexandra R. De Leon, Kevin R. Lynch, Timothy L. Macdonald
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 23) pp:7132-7136
Publication Date(Web):1 December 2010
DOI:10.1016/j.bmcl.2010.09.030
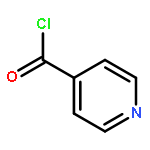
Autotaxin (ATX) is a secreted soluble enzyme that generates lysophosphatidic acid (LPA) through its lysophospholipase D activity. Because of LPA’s role in neoplastic diseases, ATX is an attractive therapeutic target due to its involvement in LPA biosynthesis. Here we describe the SAR of ATX inhibitor, VPC8a202, and apply this SAR knowledge towards developing a high potency inhibitor. We found that electron density in the pyridine region greatly influences activity of our inhibitors at ATX.
Co-reporter:Fengping Li, Mahendra D. Chordia, Tao Huang and Timothy L. Macdonald
Chemical Research in Toxicology 2009 Volume 22(Issue 1) pp:72
Publication Date(Web):December 3, 2008
DOI:10.1021/tx800152r
Nimesulide is a nonsteroidal anti-inflammatory drug (NSAID) marketed in more than 50 countries. This drug has caused rare and idiosyncratic but severe hepatotoxicity. The mechanisms associated with and factors responsible for this toxicity remain unknown. One of the nimesulide metabolites identified in human urine is 4-amino-2-phenoxy-methanesulfonanilide (M1). In the current study, we demonstrate that M1 is a stable metabolite that is highly susceptible to facile oxidation by cytochrome P450 enzymes (P450s) to form a reactive diiminoquinone intermediate (M2). Direct detection of M2 was difficult by LC-MS. However, its formation was confirmed indirectly by identification of N-acetyl-cysteine (NAC) adducts of M2. The formation of diiminoquinone M2 was P450 mediated with 2C19 and 1A2 as the two principal P450 enzymes catalyzing M1 oxidation. M1 metabolism irreversibly inhibited 2C19 but activated 1A2 in a time-dependent manner. P450 2C19 exclusively mediated further metabolism of M1 to the amino hydroxynimesulide M3 and its diiminoquinone M4. Similar to M2, M4 is also reactive and can be observed indirectly as its NAC adduct. Nucleophilic addition to diiminoquinone M2 occurs with low regioselectivity, yielding three adducts (the peak area ratio 1:0.08:12). The three regioisomers have the same m/z for [M + H]+, presumably due to nucleophilic addition at the three possible electrophilic sites (C-3, -5, and -6 positions of the sulfonaniline ring). The primary adduct, R, was derived from the attack of the nucleophile at the C-5 position of the sulfonaniline ring and was determined by MS/MS and 1H and 13C NMR analyses. The structural assignments were confirmed by chemical synthesis of the adduct R. M2 demonstrated its electrophilic reactivity by selectively alkylating human serum albumin (HSA) at the only free thiol, Cys-34. This suggests the possibility that other proteins may undergo a similar conjugation to form irreversible adducts. Under oxidizing conditions in the presence of cumene hydroperoxide (CHP), the formation of M2 was enhanced, indicating that oxidative stress may accelerate the production of reactive diiminoquinone species (M2 and M4).
Co-reporter:Frank W. Foss Jr., Thomas P. Mathews, Yugesh Kharel, Perry C. Kennedy, Ashley H. Snyder, Michael D. Davis, Kevin R. Lynch, Timothy L. Macdonald
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 16) pp:6123-6136
Publication Date(Web):15 August 2009
DOI:10.1016/j.bmc.2009.04.015
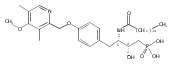
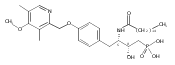
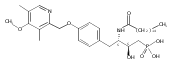
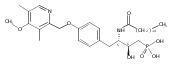
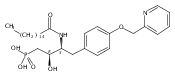
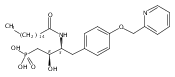
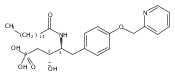
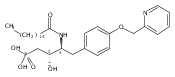
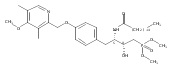
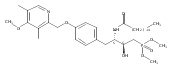
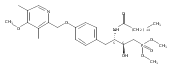
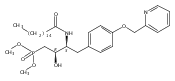
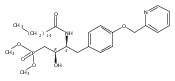
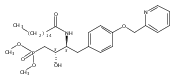
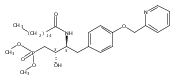
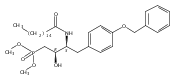
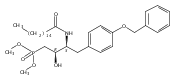
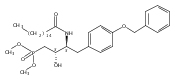
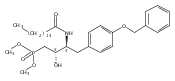
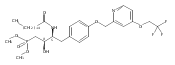
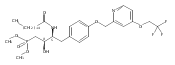
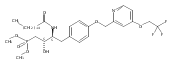
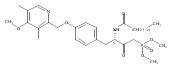
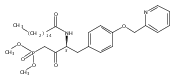
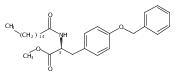
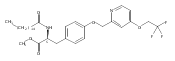
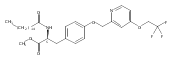
In the search for bioactive sphingosine 1-phosphate (S1P) receptor ligands, a series of 2-amino-2-heterocyclic-propanols were synthesized. These molecules were discovered to be substrates of human-sphingosine kinases 1 and 2 (SPHK1 and SPHK2). When phosphorylated, the resultant phosphates showed varied activities at the five sphingosine-1-phosphate (S1P) receptors (S1P1–5). Agonism at S1P1 was displayed in vivo by induction of lymphopenia. A stereochemical preference of the quaternary carbon was crucial for phosphorylation by the kinases and alters binding affinities at the S1P receptors. Oxazole and oxadiazole compounds are superior kinase substrates to FTY720, the prototypical prodrug immunomodulator, fingolimod (FTY720). The oxazole-derived structure was the most active for human SPHK2. Imidazole analogues were less active substrates for SPHKs, but more potent and selective agonists of the S1P1 receptor; additionally, the imidazole class of compounds rendered mice lymphopenic.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Qin Sun, Ran Zhu, Frank W. Foss Jr. and Timothy L. Macdonald
Chemical Research in Toxicology 2008 Volume 21(Issue 3) pp:711
Publication Date(Web):February 26, 2008
DOI:10.1021/tx7003085
Trovafloxacin (Trovan) is a fluoroquinolone antibiotic drug with a long half-life and broad-spectrum activity. Since its entry into the market in 1998, trovafloxacin has been associated with numerous cases of hepatotoxicity, which has limited its clinical usefulness. Trovafloxacin possesses two substructural elements that have the potential to generate reactive intermediates: a cyclopropylamine moiety and a difluoroanilino system. The results presented here describe the in vitro metabolic activation of a synthetic drug model (DM) of trovafloxacin that contains the cyclopropylamine moiety. Cyclopropylamine can be oxidized to reactive ring-opened products—a carbon-centered radical and a subsequently oxidized α,β-unsaturated aldehyde. Experiments with monoamine oxygenases, horseradish peroxidase, flavin monooxygenase 3, and cDNA-expressed P450 isoenzymes revealed that P450 1A2 oxidizes DM to a reactive α,β-unsaturated aldehyde, M1. Furthermore, myeloperoxidase (MPO) was also demonstrated to oxidize DM in the presence of chloride ion to produce M1. DM proved to be a suicide inhibitor of MPO while showing no inhibition of P450 1A2. The structure of the reactive metabolite was confirmed by LC-MS/MS analysis by comparison with a synthetic standard. M1 was further shown to react with glutathione and the related thiol nucleophile, 4-bromobenzyl mercaptan, suggesting the potential of this intermediate to react with protein nucleophiles. In summary, these data provide evidence that trovafloxacin-induced hepatotoxicity may be mediated through the oxidation of the cyclopropylamine substructure to reactive intermediates that may form covalent adducts to hepatic proteins, resulting in damage to liver tissue.
Co-reporter:Ran Zhu ; Ashley H. Snyder ; Yugesh Kharel ; Lisa Schaffter ; Qin Sun ; Perry C. Kennedy ; Kevin R. Lynch
Journal of Medicinal Chemistry 2007 Volume 50(Issue 25) pp:6428-6435
Publication Date(Web):November 10, 2007
DOI:10.1021/jm7010172
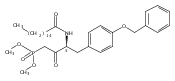
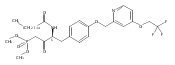
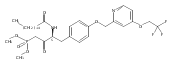
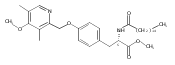
Compound 1 (FTY720, Fingolimod) represents a new generation of immunosuppressant that modulates lymphocyte trafficking by interacting with the S1P1 receptor. Compound 1 also provides a template molecule for studying the molecular biology of S1P receptors and related enzymes (kinases and phosphatases). In this study, two conformationally constrained analogues of 1 (3a and 3c) were asymmetrically synthesized in high optical purity. In vitro assessment documented that both analogues are Sphk2 substrates, their phosphorylated species are potent S1P1 receptor agonists, and 3a-P is a potent S1P3 antagonist. After oral administration in mice, both compounds evoked lymphopenia, but their duration of action differed markedly.
Co-reporter:Fengping Li, Mahendra D. Chordia, Kellie A. Woodling and Timothy L. Macdonald
Chemical Research in Toxicology 2007 Volume 20(Issue 12) pp:1854
Publication Date(Web):October 19, 2007
DOI:10.1021/tx7001417
2-Acetylbenzothiophene-S-oxide (2-ABT-S-oxide or M1) is a reactive metabolite of zileuton, a drug used in the treatment of asthma and is capable of conjugating with glutathione in vitro. Human serum albumin (HSA) is the most abundant protein in plasma and plays a critical role in detoxifying reactive oxygen species. The current research is focused on understanding the interaction between M1 and HSA. The stability studies revealed the half-life of M1 to be about 0.85 h in HSA, 1.82 h in human plasma, and 4.48 h in phosphate-buffered saline (PBS) as determined by first-order approximation. The alkylation rate constant k for HSA was 20 M−1 min−1. After quenching with acetonitrile, the half-life of M1 did not change significantly, indicating that M1 is covalently bound to HSA. LC–MS and LC–MS/MS analysis of human plasma revealed the M1 alkylated peptide P (m/z 870) formed by HSA conjugation and concomitant water elimination. The specific amino acid on HSA bound to M1 was identified as Cys-34. This alkylation is observed to be concentration- and incubation-time-dependent in human plasma. HSA oxidized by N,N′-diacetyl-l-cystine exhibits a compromised ability of HSA to react with M1. The alkylated HSA diminished the binding affinity for warfarin. Furthermore, the alkylation was found to be irreversible in the dialysis experiment. In addition, M1 decomposes to 2-ABT in the presence of HSA, presumably acting as an oxidant. The formation of 2-ABT in the incubation and the self-condensation of M1 in PBS indicate that the alkylation of Cys-34 is only one of a number of reactions that occur in the presence of HSA. Irreversible protein modification may potentially lead to a loss of its function. HSA irreversible alkylation represents a model for other proteins to be potentially toxic and thus may help explain zileuton hepatotoxicity.
Co-reporter:Ran Zhu, Cynthia R. Frazier, Joel Linden, Timothy L. Macdonald
Bioorganic & Medicinal Chemistry Letters 2006 Volume 16(Issue 9) pp:2416-2418
Publication Date(Web):1 May 2006
DOI:10.1016/j.bmcl.2006.01.110
A series of N6-ethyl-2-alkynyl NECA (5′-N-ethylcarboxamidoadenosine) analogs were synthesized and their binding affinity with the four human adenosine receptors was evaluated. One of the compounds ZR1121 shows high affinity with hA3 receptor and its selectivity over hA1 receptor is 1–2 log orders greater than IB-MECA or Cl-IB-MECA, the currently employed selective A3 agonists.A new adenosine analogue ZR1121 is reported. Compared with currently widely used hA3 agonists IB MECA and Cl-IB MECA, this compound has similar activity and about 100 times higher hA3/hA1 selectivity.
Co-reporter:Jeremy J. Clemens, Michael D. Davis, Kevin R. Lynch, Timothy L. Macdonald
Bioorganic & Medicinal Chemistry Letters 2003 Volume 13(Issue 20) pp:3401-3404
Publication Date(Web):20 October 2003
DOI:10.1016/S0960-894X(03)00812-6
Sphingosine-1-phosphate (S1P) is a biologically active lysophospholipid with the capacity to induce a broad range of cellular responses via its interaction with the S1P family of G-protein coupled receptors. This report describes the synthesis of several potent S1P receptor agonists. For instance, compound 9c displayed an EC50=8.6 nM at the S1P1 receptor using a [γ-35S]GTP binding assay as compared to an EC50=4.5 nM for the endogenous ligand. We also report the effects associated with introduction of a phenyl ring between the ‘linker’ and ‘lipophilic tail’ regions of the analogues, for example total loss of activity at S1P2 and increased agonism at S1P5.We report the synthesis and potencies of several novel S1P receptor agonists.
Co-reporter:Thomas A Miller, Jagadananda Ghosh, Charles E Myers, Timothy L Macdonald
Bioorganic & Medicinal Chemistry Letters 2000 Volume 10(Issue 17) pp:1913-1916
Publication Date(Web):September 2000
DOI:10.1016/S0960-894X(00)00370-X
The synthesis and assessment of the mitogenic properties of 5-HETE congeners are reported. These studies represent an effort to develop a structure–activity profile for ligands of the 5-HETE/5-oxoETE G-protein coupled receptor(s). Many of these agents possess mitogenic activity that equals or exceeds that of racemic 5-HETE family constituents in prostate cancer cell lines.
.jpg)




![2-[1-amino-2-[(2-methylpropan-2-yl)oxy]-2-oxoethyl]benzoic acid](http://img.cochemist.com/ccimg/669800/669713-61-5.png)
![2-[1-amino-2-[(2-methylpropan-2-yl)oxy]-2-oxoethyl]benzoic acid](http://img.cochemist.com/ccimg/669800/669713-61-5_b.png)
![Benzoic acid, 4-methoxy-2-[[(trifluoromethyl)sulfonyl]oxy]-, methyl ester](http://img.cochemist.com/ccimg/216800/216768-18-2.png)
![Benzoic acid, 4-methoxy-2-[[(trifluoromethyl)sulfonyl]oxy]-, methyl ester](http://img.cochemist.com/ccimg/216800/216768-18-2_b.png)




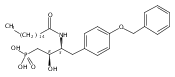
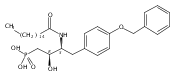
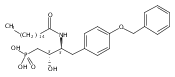
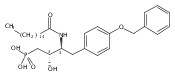

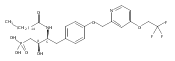
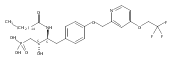
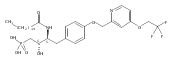
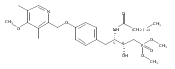




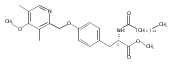
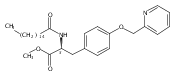
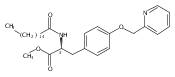
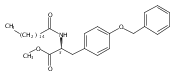

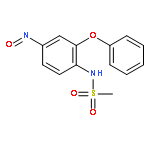
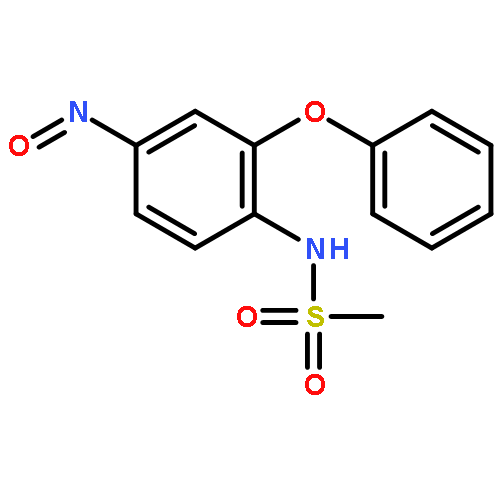


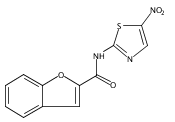
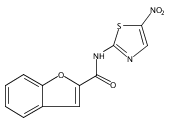
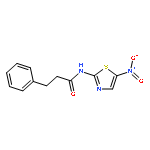
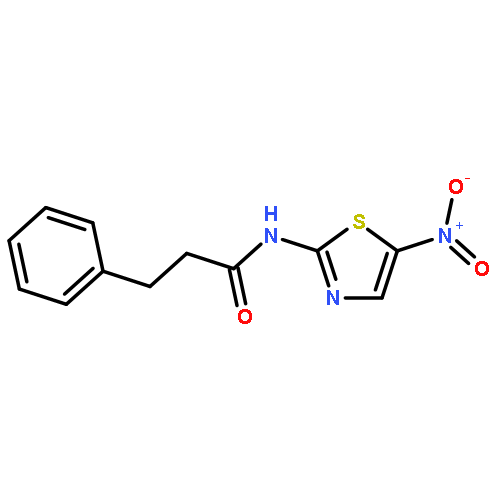
![Phenol,4-[[4-(4-chlorophenyl)-2-thiazolyl]amino]-](http://img.cochemist.com/ccimg/312700/312636-16-1.png)
![Phenol,4-[[4-(4-chlorophenyl)-2-thiazolyl]amino]-](http://img.cochemist.com/ccimg/312700/312636-16-1_b.png)




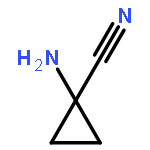
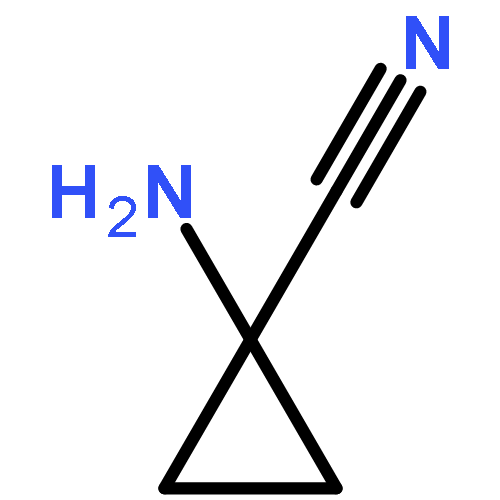
![5H-Imidazo[2,1-b][1,3]oxazine, 6,7-dihydro-2-nitro-6-[[4-(trifluoromethoxy)phenyl]methoxy]-, (6S)-](http://img.cochemist.com/ccimg/187300/187235-37-6.png)
![5H-Imidazo[2,1-b][1,3]oxazine, 6,7-dihydro-2-nitro-6-[[4-(trifluoromethoxy)phenyl]methoxy]-, (6S)-](http://img.cochemist.com/ccimg/187300/187235-37-6_b.png)
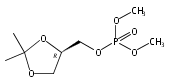


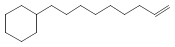














![Carbamic acid,N-[(1R)-1-cyanoethyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/101000/100927-09-1.png)
![Carbamic acid,N-[(1R)-1-cyanoethyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/101000/100927-09-1_b.png)

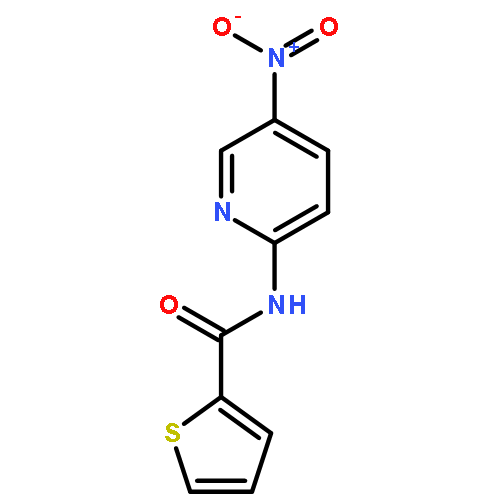
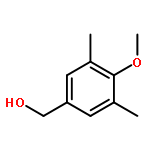
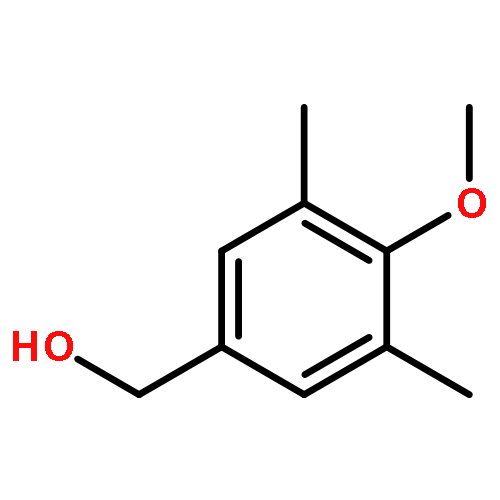
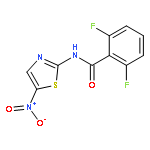

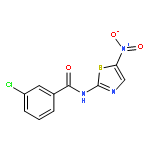
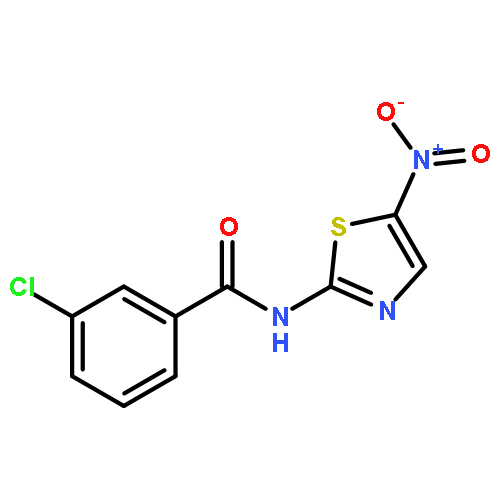
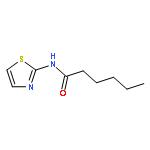
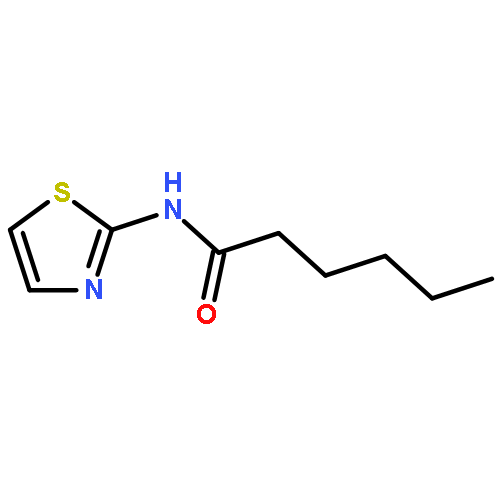

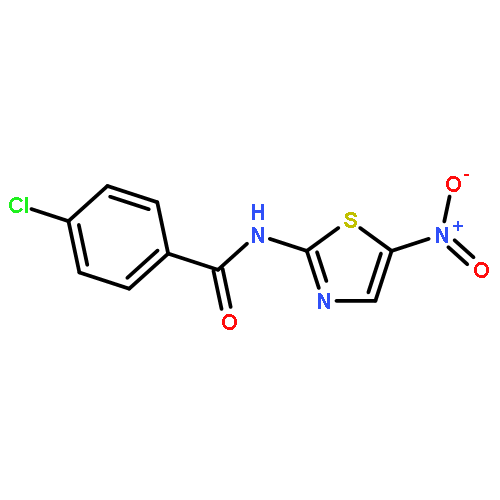
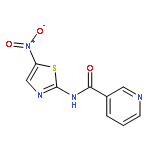
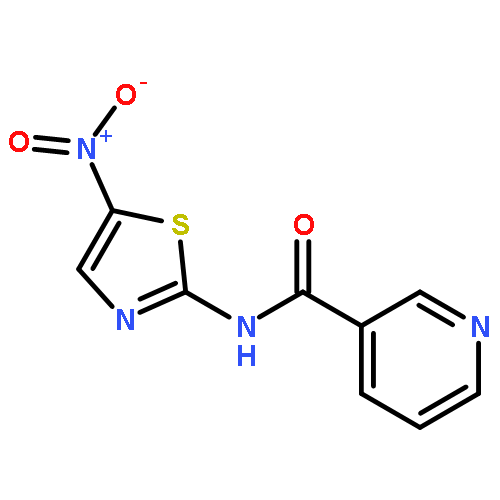
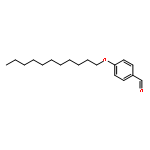

![[2,3'-Bithiophene]-5-carboxylic acid](http://img.cochemist.com/ccimg/60200/60141-31-3.png)
![[2,3'-Bithiophene]-5-carboxylic acid](http://img.cochemist.com/ccimg/60200/60141-31-3_b.png)























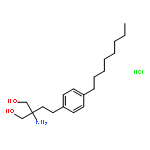
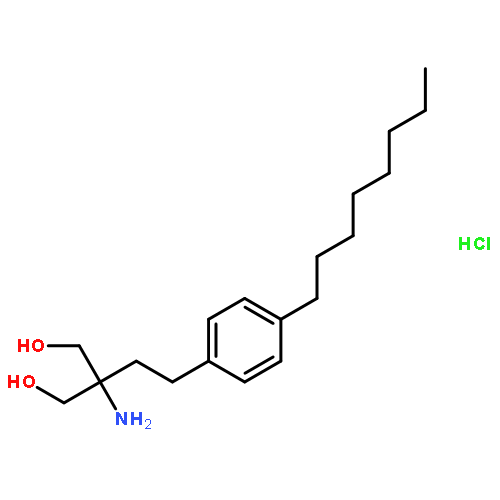






![1,3-Propanediol,2-amino-2-[2-(4-octylphenyl)ethyl]-](http://img.cochemist.com/ccimg/162400/162359-55-9.png)
![1,3-Propanediol,2-amino-2-[2-(4-octylphenyl)ethyl]-](http://img.cochemist.com/ccimg/162400/162359-55-9_b.png)