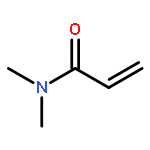
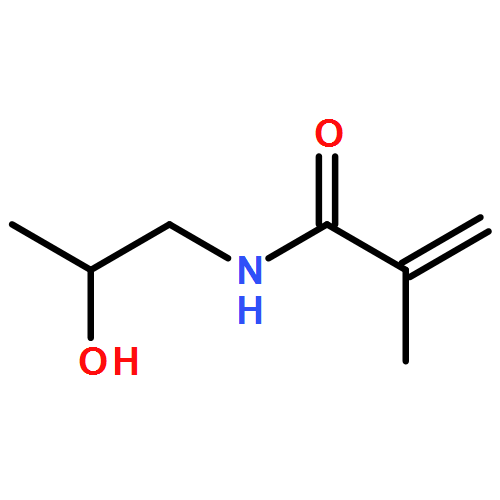
Co-reporter:Megan R. Hill, Elise Guégain, Johanna Tran, C. Adrian Figg, Andrew C. Turner, Julien Nicolas, and Brent S. Sumerlin
ACS Macro Letters October 17, 2017 Volume 6(Issue 10) pp:1071-1071
Publication Date(Web):September 15, 2017
DOI:10.1021/acsmacrolett.7b00572
Radical copolymerization of donor–acceptor (D-A) monomer pairs has served as a versatile platform for the development of alternating copolymers. However, due to the use of conventional radical polymerization, the resulting copolymers have generally been limited to nondegradable vinyl polymers. By combining radical D-A copolymerization with radical ring-opening polymerization (rROP), we have synthesized an alternating copolymer with a high incorporation of degradable backbone units. Copolymerization of N-ethyl maleimide (NEtMI) with the cyclic ketene acetal (CKA) 2-methylene-4-phenyl-1,3-dioxolane (MPDL) was demonstrated to proceed in an alternating fashion, and controlled polymerization was achieved using reversible addition–fragmentation chain transfer (RAFT) polymerization. Spontaneous copolymerization, in the absence of an exogenous initiating source, occurred when the mixture of monomers was heated, presumably due to the large electron disparity between the comonomers. Chain-extension with styrene afforded well-defined P(MPDL-alt-NEtMI)-b-polystyrene copolymers, and degradation of the homopolymers and block copolymers showed complete breakdown of the alternating copolymer.
Co-reporter:Eva Blasco, Michael B. Sims, Anja S. Goldmann, Brent S. Sumerlin, and Christopher Barner-Kowollik
Macromolecules July 25, 2017 Volume 50(Issue 14) pp:5215-5215
Publication Date(Web):June 26, 2017
DOI:10.1021/acs.macromol.7b00465
The translation of small molecule chemistries into efficient methodologies for polymer functionalization spans several decades, enabling critical advances in soft matter materials synthesis with tailored and adaptive property profiles. The present Perspective explores—based on selected examples—50 years of innovation in polymer functionalization chemistries. These span a diverse set of chemistries based on activated esters, thiol–ene/yne processes, nucleophilic systems based on isocyanates, reactions driven by the formation of imines and oximes, ring-opening processes, cycloadditions, and—in a recent renaissance—multicomponent reactions. In addition, a wide variety of chain types and architectures have been modified based on the above chemistries, often with exquisite chemical control, highlighted by key examples. We conclude our journey through polymer functionalization with the—in our view—most critically required advances that have the potential to move from “science fiction” to “science fact”.
Co-reporter:C. Adrian Figg, R. Nicholas Carmean, Kyle C. Bentz, Soma Mukherjee, Daniel A. Savin, and Brent S. Sumerlin
Macromolecules 2017 Volume 50(Issue 3) pp:
Publication Date(Web):January 23, 2017
DOI:10.1021/acs.macromol.6b02754
Hydrophobicity inherently affects a solutes behavior in water, yet how polymer chain hydrophobicity impacts aggregate morphology during solution self-assembly and reorganization is largely overlooked. As polymer and nanoparticle syntheses are easily achieved, the resultant nanoparticle architectures are usually attributed to chain topology and overall degree of polymerization, bypassing how the chains may interact with water during/after self-assembly to elicit morphology changes. Herein, we demonstrate how block copolymer hydrophobicity allows control over aggregate morphology in water and leads to remarkable control over the length of polymeric nanoparticle worms. Polymerization-induced self-assembly facilitated nanoparticle synthesis through simultaneous polymerization, self-assembly, and chain reorganization during a block copolymer chain extension from a hydrophilic poly(N,N-dimethylacrylamide) macro-chain-transfer agent with diacetone acrylamide and N,N-dimethylacrylamide. Slight variations in the monomer feed ratio dictated the block copolymer chain composition and were proposed to alter aggregate thermodynamics. Micelles, worms, and vesicles were synthesized, and the highest level of control over worm elongation attained during a polymerization is reported, simply due to the polymer chain hydrophobicity.
Co-reporter:R. Nicholas Carmean, Troy E. Becker, Michael B. Sims, Brent S. Sumerlin
Chem 2017 Volume 2, Issue 1(Volume 2, Issue 1) pp:
Publication Date(Web):12 January 2017
DOI:10.1016/j.chempr.2016.12.007
•Aqueous, catalyst-free route leads to polymers with unprecedented chain lengths•Polymerization control is mediated by mild UV or sunlight irradiation•Synthesis of UHMW block copolymers could facilitate access to new advanced materialsMaterials derived from ultra-high-molecular-weight (UHMW) polymers offer unrivaled mechanical strength but are often limited in composition and architecture. Although advances in living polymerizations, especially reversible-deactivation radical polymerization (RDRP), have enabled the design of well-defined polymers with controlled molecular weights and architectures, most methods enable control only up to modest molecular weights, despite the need for new robust materials. An ability to target complex UHMW polymers via RDRP could allow an unprecedented opportunity to investigate important fundamental principles in self-assembly behavior and phase segregation. Herein, we describe catalyst-free photopolymerization conditions that facilitate the synthesis of UHMW polymers in environmentally friendly aqueous solvents to achieve near-quantitative monomer conversion by requiring only a readily available and low-energy light source or, in some cases, only sunlight.Relying solely on mild UV irradiation of thiocarbonylthio compounds in the presence of vinyl monomers, a new avenue to well-defined ultra-high-molecular-weight (UHMW) polymers has been developed. Through the use of aqueous conditions, well-controlled UHMW polymers that are unprecedented for controlled radical polymerizations have been achieved. This photomediated polymerization approach reaches number-average molecular weights in excess of 8.00 × 106 g/mol with degrees of polymerization above 85,000, making these, to our knowledge, the highest-molecular-weight polymers ever achieved via reversible-deactivation radical polymerization. In many cases, well-defined UHMW polymers can be obtained in minutes. The utility of the technique is further demonstrated through the synthesis of block copolymers, enabling access to a new field of well-defined UHMW materials.Download high-res image (131KB)Download full-size image
Co-reporter:Yuqiong Dai;Hao Sun;Sunirmal Pal;Yunlu Zhang;Sangwoo Park;Christopher P. Kabb;Wei David Wei
Chemical Science (2010-Present) 2017 vol. 8(Issue 3) pp:1815-1821
Publication Date(Web):2017/02/28
DOI:10.1039/C6SC04650A
Responsive systems sensitive to near-infrared (NIR) light are promising for triggered release due to efficient deep tissue penetration of NIR irradiation relative to higher energy sources (e.g., UV), allowing for spatiotemporal control over triggering events with minimal potential for tissue damage. Herein, we report star polymers containing thermally-labile azo linkages that dissociate during conventional heating or during localized heating via the photothermal effect upon NIR irradiation. Controlled release during conventional heating was investigated for the star polymers loaded with a model dye, with negligible release being observed at 25 °C and >80% release at 90 °C. Star polymers co-loaded with NIR-responsive indocyanine green showed rapid dye release upon NIR irradiation (λ ≥ 715 nm) due to the photothermally-induced degradation of azo linkages within the cores of the star polymers. This approach provides access to a new class of delivery and release systems that can be triggered by noninvasive external stimulation.
Co-reporter:Yuan Liu;Weijia Hou;Hao Sun;Cheng Cui;Liqin Zhang;Ying Jiang;Yongxiang Wu;Yanyue Wang;Juan Li;Qiaoling Liu;Weihong Tan
Chemical Science (2010-Present) 2017 vol. 8(Issue 9) pp:6182-6187
Publication Date(Web):2017/08/21
DOI:10.1039/C7SC01447C
Bioconjugation based on crosslinking primary amines to carboxylic acid groups has found broad applications in protein modification, drug development, and nanomaterial functionalization. However, proteins, which are made up of amino acids, typically give nonselective bioconjugation when using primary amine-based crosslinking. In order to control protein orientation and activity after conjugation, selective bioconjugation is desirable. We herein report an efficient and cysteine-selective thiol–ene click reaction-based bioconjugation strategy using colloidal nanoparticles. The resulting thiol–ene based aptamer and enzyme nanoconjugates demonstrated excellent target binding ability and enzymatic activity, respectively. Thus, thiol–ene click chemistry can provide a stable and robust crosslinker in a biocompatible manner for bioconjugation of any thiol-containing biomolecule with nanomaterials. This will open more opportunities for applications of thiol–ene reactions and functional colloidal nanoparticles in chemical biology.
Co-reporter:R. Nicholas Carmean, C. Adrian Figg, Georg M. Scheutz, Tomohiro Kubo, and Brent S. Sumerlin
ACS Macro Letters 2017 Volume 6(Issue 2) pp:
Publication Date(Web):February 9, 2017
DOI:10.1021/acsmacrolett.7b00038
An initiator- and catalyst-free method for polymer end-group modification has been designed. Under long-wave ultraviolet irradiation, polymers with thiocarbonylthio end groups undergo photolytic cleavage to reveal an active macroradical capable of irreversible termination with a suitable hydrogen source. This straightforward method was successfully demonstrated by the removal of a range of end groups that commonly result from reversible addition–fragmentation chain transfer or photoiniferter polymerizations, including trithiocarbonate, dithiobenzoate, xanthate, and dithiocarbamate mediating agents. This strategy proved efficient for polymers derived from acrylamido, acrylic, methacrylic, styrenic, and vinylpyrrolidone monomers.
Co-reporter:Tomohiro Kubo;Kyle C. Bentz;Kristin C. Powell;C. Adrian Figg;Jeremy L. Swartz;Maxym Tansky;Anuj Chauhan;Daniel A. Savin
Polymer Chemistry (2010-Present) 2017 vol. 8(Issue 39) pp:6028-6032
Publication Date(Web):2017/10/10
DOI:10.1039/C7PY01585B
Herein, we demonstrate a concise and modular preparation of homopolymers with both a hydrophilic and hydrophobic group on each monomer unit. Successive and chemoselective nucleophilic aromatic substitution of polymers derived from 2,4,6-trichloro-1,3,5-triazine allowed the step-wise installation of hydrophiles and hydrophobes and facilitated the systematic investigation of structure–property relationships for this new class of amphiphilic homopolymers.
Co-reporter:Jawaher A. Alfurhood;Hao Sun;Christopher P. Kabb;Bryan S. Tucker;James H. Matthews;Hendrik Luesch
Polymer Chemistry (2010-Present) 2017 vol. 8(Issue 34) pp:4983-4987
Publication Date(Web):2017/08/30
DOI:10.1039/C7PY00196G
We report nanoassemblies based on block copolymers of N-(2-hydroxypropyl)methacrylamide (HPMA) in which drug cleavage enhances the biological compatibility of the original polymer carrier by regeneration of HPMA units. Drug release via ester hydrolysis suggests this approach offers potential for stimuli-responsive drug delivery under acidic conditions.
Co-reporter:Charles P. Easterling;Tomohiro Kubo;Zachary M. Orr;Gail E. Fanucci
Chemical Science (2010-Present) 2017 vol. 8(Issue 11) pp:7705-7709
Publication Date(Web):2017/10/23
DOI:10.1039/C7SC02574B
The direct transformation of commercially available commodity polyacrylates into value-added materials was achieved. We demonstrate how 1,5,7-triazabicyclo[4.4.0]dec-5-ene, serving as a nucleophilic catalyst, can be used to catalyze acyl substitution reactions of acrylic polymers in the presence of alcohol and amine nucleophiles. Furthermore, we found that organocatalytic transesterification exhibits high selectivity towards sterically unhindered esters, thus providing a new route towards site-selective acyl substitution of macromolecular materials. Combining this methodology with reversible-deactivation radical polymerization (RDRP) techniques such as reversible addition–fragmentation chain-transfer (RAFT) polymerization allowed for the precise functionalization of sterically-differentiated acrylic copolymers and polymeric chain ends. We envision this approach to expedite functional polymer synthesis and provide access to functional macromolecules prepared from inexpensive, hydrolytically-stable polymeric precursors.
Co-reporter:C. Adrian Figg;Ashton N. Bartley;Tomohiro Kubo;Bryan S. Tucker;Ronald K. Castellano
Polymer Chemistry (2010-Present) 2017 vol. 8(Issue 16) pp:2457-2461
Publication Date(Web):2017/04/18
DOI:10.1039/C7PY00225D
This report describes the efficient preparation of ω,ω-heterodifunctionalized polymers and polymer bioconjugates under mild conditions using a recently introduced reagent, benzotrifuranone (BTF). Demonstrated is how BTF enables introduction of differentially “clickable” functional groups (e.g., alkenes and alkynes) to monomethyl ether poly(ethylene glycol) amine at ambient temperature using near-stoichiometric amounts of reagents, in contrast to conventional polymer heterofunctionalization approaches that may require high temperatures, significant excesses of reagents, and/or numerous synthetic steps. Showcasing the methodology is facile access to an ω,ω-heterodifunctional polymer bearing a fluorescent (coumarin) dye and biotin. Found, in addition to avidin binding by the polymer bioconjugate, is the unexpected disruption of avidin tetramer formation.
Co-reporter:William L. A. Brooks and Brent S. Sumerlin
Chemical Reviews 2016 Volume 116(Issue 3) pp:1375
Publication Date(Web):September 14, 2015
DOI:10.1021/acs.chemrev.5b00300
Co-reporter:Jawaher A. Alfurhood, Hao Sun, Patricia R. Bachler and Brent S. Sumerlin
Polymer Chemistry 2016 vol. 7(Issue 11) pp:2099-2104
Publication Date(Web):25 Feb 2016
DOI:10.1039/C6PY00111D
We report the first synthesis of hyperbranched poly(N-(2-hydroxypropyl) methacrylamide) (HB-PHPMA) using reversible addition–fragmentation chain transfer (RAFT) self-condensing vinyl polymerization (SCVP). The synthesis of these complex, well-defined architectures involved the copolymerization of HPMA with a chain transfer monomer (CTM). The polymerization kinetics, as well as the effects of initiator concentration and the ratio of monomer to CTM were studied. The resulting polymers show high molecular weights and controlled branching frequencies. Due to their inherent amphiphilic nature, these hyperbranched structures self-assemble into aggregates in water. Additionally, we determined the cloud point of the HB-PHPMA to be in the range of sub-ambient temperature to 40 °C. This approach provides access to a new class of thermoresponsive PHPMA polymers with potential to be used in drug delivery and other biological applications.
Co-reporter:Patricia R. Bachler, Kaitlyn E. Forry, Chelsea A. Sparks, Michael D. Schulz, Kenneth B. Wagener and Brent S. Sumerlin
Polymer Chemistry 2016 vol. 7(Issue 25) pp:4155-4159
Publication Date(Web):06 Jun 2016
DOI:10.1039/C6PY00819D
Modular segmented hyperbranched polymers, amenable to facile post-polymerization functionalization, were created via two distinct approaches. Self-condensing vinyl polymerization via reversible addition–fragmentation chain transfer (RAFT) polymerization and RAFT polymerization with a divinyl comonomer were employed to create well-defined highly branched materials containing activated esters amenable to highly efficient functionalization in a modular manner.
Co-reporter:Soma Mukherjee, William. L. A. Brooks, Yuqiong Dai and Brent S. Sumerlin
Polymer Chemistry 2016 vol. 7(Issue 10) pp:1971-1978
Publication Date(Web):17 Feb 2016
DOI:10.1039/C5PY02046H
Two sets of reversible covalent linkages distributed in series along a polymer backbone were used to prepare a new class of doubly-dynamic-covalent polymers capable of reversibly dissociating via two distinct pathways. These self-repairable linear polymers were prepared via step-growth Diels–Alder polymerization of an AB monomer that contained furfuryl- and maleimido groups linked by an oxime bond. Both the oxime and oxanorbornene links lent orthogonally reversible character to the polymer backbone. The sequentially distributed oxime bonds in the polymer were capable of dynamic oxime exchange in the presence of a competitive monofunctional alkoxyamine under acidic conditions, while the oxanorbornene linkages were susceptible to cleavage via retro-Diels–Alder reactions at elevated temperatures and recombination upon cooling. The self-healing or reversible nature of the dynamic-covalent oxime bonds and oxanorbornene links has potential for designing stimuli-responsive self-repairable materials for sensors and drug delivery applications.
Co-reporter:Jawaher A. Alfurhood, Patricia R. Bachler and Brent S. Sumerlin
Polymer Chemistry 2016 vol. 7(Issue 20) pp:3361-3369
Publication Date(Web):28 Apr 2016
DOI:10.1039/C6PY00571C
RAFT-mediated self-condensing vinyl polymerization is a promising synthetic tool to create well-defined hyperbranched polymers. The functional group tolerance of RAFT, as well as the inherent ability to install readily functionalizable end groups, allow for formation of highly functionalized materials with unique properties. Due to the controlled nature of RAFT, it is possible to synthesize polymers with predetermined branching frequencies, resulting in materials with tunable properties and readily tailored macromolecular structure. This minireview gives an overview of this emerging field, emphasizing structural variety, monomer functionality, and post-polymerization modifications that have been employed to prepare previously inaccessible materials.
Co-reporter:Hao Sun, Daniel J. Dobbins, Yuqiong Dai, Christopher P. Kabb, Shijian Wu, Jawaher A. Alfurhood, Carlos Rinaldi, and Brent S. Sumerlin
ACS Macro Letters 2016 Volume 5(Issue 6) pp:688
Publication Date(Web):May 19, 2016
DOI:10.1021/acsmacrolett.6b00327
The design and synthesis of a new class of thermally-labile poly(β-thioester)s is reported. Aliphatic azo linkages were incorporated into the main chain of the polymers to allow for degradation to lower molecular weights upon heating. These polymers displayed a temperature-dependent degradation profile with a significant increase in decomposition rate as the temperature was raised from 60 to 95 °C. This approach was further extended to prepare amphiphilic triblock copolymers containing poly(β-thioester)s and poly(ethylene glycol) (PEG). The resulting block copolymers were capable of self-assembly into micelles in water. Moreover, the assembled nanoparticles underwent dissociation as a result of exposure to heat.
Co-reporter:Sunirmal Pal, William L. A. Brooks, Daniel J. Dobbins, and Brent S. Sumerlin
Macromolecules 2016 Volume 49(Issue 24) pp:9396-9405
Publication Date(Web):December 12, 2016
DOI:10.1021/acs.macromol.6b02079
Radical copolymerization of divinyl monomers in the presence of chain transfer agents leads to soluble hyperbranched polymers. In this work, hyperbranched poly(poly(ethylene glycol) methyl ether methacrylate) (PPEGMA) with degradable cross-linker branch points derived from glucarodilactone methacrylate was prepared via reversible addition–fragmentation chain transfer (RAFT) polymerization to provide insight into hyperbranch formation during copolymerizations of multiolefinic compounds. The number-average molecular weight of the polymers increased nonlinearly with monomer conversion, implying that the incorporation of the divinyl cross-linker led to chain branching and a rapid increase in molecular weight at high conversion. The degree of branching was varied by controlling the feed ratio of monomer to cross-linker to chain transfer agent. Hydrolytic degradation of the sugar-derived dilactone branch points was examined under acidic, neutral, and basic aqueous conditions. To provide fundamental insight into the growth of primary chains during RAFT polymerizations of multiolefinic compounds, the resulting hyperbranched polymers were subjected to cross-link cleavage to obtain linear polymers. The molecular weights of the resulting polymer segments were similar to the theoretical molecular weights expected for linear analogues prepared with similar ratios of monomer to RAFT agent. Not only does this approach lead to new examples of degradable polymers with complex architectures, but also to important mechanistic insights into hyperbranch formation via polymerization of multiolefinic compounds.
Co-reporter:R. Nicholas Carmean;C. Adrian Figg;Troy E. Becker ; Brent S. Sumerlin
Angewandte Chemie International Edition 2016 Volume 55( Issue 30) pp:8624-8629
Publication Date(Web):
DOI:10.1002/anie.201603129
Abstract
A biphasic one-pot polymerization method enables the preparation of block copolymers from monomers with similar and competitive reactivities without the addition of external materials. AB diblock copolymers were prepared by encapsulating a frozen solution of monomer B on the bottom of a reaction vessel, while the solution polymerization of monomer A was conducted in a liquid layer above. Physical separation between the solid and liquid phases permitted only homopolymerization of monomer A until heating above the melting point of the lower phase, which released monomer B, allowing the addition of the second block to occur. The triggered release of monomer B allowed for chain extension without additional deoxygenation steps or exogenous monomer addition. A method for the closed (i.e., without addition of external reagents) one-pot synthesis of block copolymers with conventional glassware using straightforward experimental techniques has thus been developed.
Co-reporter:R. Nicholas Carmean;C. Adrian Figg;Troy E. Becker ; Brent S. Sumerlin
Angewandte Chemie 2016 Volume 128( Issue 30) pp:8766-8771
Publication Date(Web):
DOI:10.1002/ange.201603129
Abstract
A biphasic one-pot polymerization method enables the preparation of block copolymers from monomers with similar and competitive reactivities without the addition of external materials. AB diblock copolymers were prepared by encapsulating a frozen solution of monomer B on the bottom of a reaction vessel, while the solution polymerization of monomer A was conducted in a liquid layer above. Physical separation between the solid and liquid phases permitted only homopolymerization of monomer A until heating above the melting point of the lower phase, which released monomer B, allowing the addition of the second block to occur. The triggered release of monomer B allowed for chain extension without additional deoxygenation steps or exogenous monomer addition. A method for the closed (i.e., without addition of external reagents) one-pot synthesis of block copolymers with conventional glassware using straightforward experimental techniques has thus been developed.
Co-reporter:Tomohiro Kubo, C. Adrian Figg, Jeremy L. Swartz, William L. A. Brooks, and Brent S. Sumerlin
Macromolecules 2016 Volume 49(Issue 6) pp:2077-2084
Publication Date(Web):March 3, 2016
DOI:10.1021/acs.macromol.6b00181
Despite recent progress for efficient and precise synthesis of functional polymers, few reports describe postpolymerization functionalization strategies for the controlled incorporation of multiple pendent groups per repeat unit. Cyanuric chloride, or 2,4,6-trichloro-1,3,5-triazine (TCT), offers a facile method to introduce distinct pendent functionalities. An acrylamide monomer containing a triazine ring with two electrophilic sites was prepared from TCT and polymerized via reversible addition–fragmentation chain transfer (RAFT) polymerization. Subsequent nucleophilic aromatic substitution reactions using a combination of amine and thiol nucleophiles introduced two functionalities into each repeat unit of the RAFT-derived polymers. The success of each postpolymerization modification reaction was confirmed by 1H NMR spectroscopy and size-exclusion chromatography, and small molecule model studies corroborated the high chemoselectivity of the nucleophilic aromatic substitution reactions. This postpolymerization modification strategy based on TCT enables a facile and efficient synthesis of multifunctional homopolymers.
Co-reporter:Christopher P. Kabb, R. Nicholas Carmean and Brent S. Sumerlin
Chemical Science 2015 vol. 6(Issue 10) pp:5662-5669
Publication Date(Web):10 Jul 2015
DOI:10.1039/C5SC01535A
The surface-localized hyperthermia of gold nanoparticles under microwave irradiation was examined. Gold nanoparticles with a hydrodynamic diameter of ∼6 nm stabilized by polymeric “thermometers” were used to gather information on the extent of heating as well as its spatial confinements. Reversible addition–fragmentation chain transfer polymerization was employed to synthesize well-defined, functional polymers of predetermined molecular weights, allowing for estimation of the distance between the nanoparticle surface and the polymer chain end. The polymers were conjugated with a fluorescent dye separated by a thermally-labile azo linkage, and these polymeric ligands were bound to gold nanoparticles via gold–thiolate bonds. Conventional heating experiments elucidated the relationship between temperature and the extent of dye release from the gold nanoparticle using fluorescence spectroscopy. The local temperature increase experienced under microwave irradiation was calculated using the same methodology. This approach indicated the temperature near the surface of the nanoparticle was nearly 70 °C higher than the bulk solution temperature, but decreased rapidly with distance, with no noticeable temperature increase when the azo linkage was approximately 2 nm away.
Co-reporter:C. Adrian Figg, Alexandre Simula, Kalkidan A. Gebre, Bryan S. Tucker, David M. Haddleton and Brent S. Sumerlin
Chemical Science 2015 vol. 6(Issue 2) pp:1230-1236
Publication Date(Web):14 Nov 2014
DOI:10.1039/C4SC03334E
Polymerization-induced self-assembly (PISA) is a versatile technique to achieve a wide range of polymeric nanoparticle morphologies. Most previous examples of self-assembled soft nanoparticle synthesis by PISA rely on a growing solvophobic polymer block that leads to changes in nanoparticle architecture during polymerization in a selective solvent. However, synthesis of block copolymers with a growing stimuli-responsive block to form various nanoparticle shapes has yet to be reported. This new concept using thermoresponsive polymers is termed polymerization-induced thermal self-assembly (PITSA). A reversible addition-fragmentation chain transfer (RAFT) polymerization of N-isopropylacrylamide from a hydrophilic chain transfer agent composed of N,N-dimethylacrylamide and acrylic acid was carried out in water above the known lower critical solution temperature (LCST) of poly(N-isopropylacrylamide) (PNIPAm). After reaching a certain chain length, the growing PNIPAm self-assembled, as induced by the LCST, into block copolymer aggregates within which dispersion polymerization continued. To characterize the nanoparticles at ambient temperatures without their dissolution, the particles were crosslinked immediately following polymerization at elevated temperatures via the reaction of the acid groups with a diamine in the presence of a carbodiimide. Size exclusion chromatography was used to evaluate the unimer molecular weight distributions and reaction kinetics. Dynamic light scattering and transmission electron microscopy provided insight into the size and morphologies of the nanoparticles. The resulting block copolymers formed polymeric nanoparticles with a range of morphologies (e.g., micelles, worms, and vesicles), which were a function of the PNIPAm block length.
Co-reporter:Mingsheng Chen, Shaun P. Jensen, Megan R. Hill, Gloria Moore, Zhenli He and Brent S. Sumerlin
Chemical Communications 2015 vol. 51(Issue 47) pp:9694-9697
Publication Date(Web):15 May 2015
DOI:10.1039/C5CC02726H
While polymeric nanocarriers are widely used in medicine for controlled release and site-specific delivery, few reports have applied such delivery methods within agriculture, despite the urgent need for specific delivery of pesticides and nutrients. We report the synthesis of stimuli-responsive and biodegradable polymeric nanocarriers designed for delivery to the phloem of plants and describe methods employed to evaluate their toxicity in plant cells.
Co-reporter:Qijing Chen, Megan R. Hill, William L. A. Brooks, Anqi Zhu, Brent S. Sumerlin, and Zesheng An
ACS Applied Materials & Interfaces 2015 Volume 7(Issue 39) pp:21668
Publication Date(Web):September 24, 2015
DOI:10.1021/acsami.5b07456
We report emulsion studies using poly(vinylphenyl boronic acid) (PVPBA) linear homopolymer as an effective emulsifier and gelator. Two stabilizing regimes were identified depending on the pH of PVPBA aqueous solutions, i.e., emulsions stabilized by the hompolymer nanoparticles (Pickering emulsions) at pH < pKa and emulsions stabilized by the homopolymer unimers at pH > pKa. In both cases, gelled emulsions were obtained from medium to high internal phase volume fractions with the unimers exhibiting more effective emulsification and gelling properties. Hydrogen bonding between the boronic acid units is proposed to account for the high strength of the emulsions. The emulsions were shown to be pH- and sugar-responsive. Finally, the stable emulsions were used as templates to directly prepare PVPBA macroporous materials and to fabricate multilayered capsules. This remarkable observation that a simple homopolymer can serve as an effective emulsifier and gelator may dramatically extend the scope of potential emulsifiers and inspire further research in the design of new types of efficient emulsifying agents.Keywords: emulsions; gels; H bonding; interfaces; polymers
Co-reporter:Christopher C. Deng, William L.A. Brooks, Khalil A. Abboud, and Brent S. Sumerlin
ACS Macro Letters 2015 Volume 4(Issue 2) pp:220
Publication Date(Web):January 26, 2015
DOI:10.1021/acsmacrolett.5b00018
This report describes the synthesis and characterization of boronate ester-cross-linked hydrogels capable of self-healing behavior at neutral and acidic pH. This atypically wide pH range over which healing behavior is observed was achieved through the use of an intramolecular coordinating boronic acid monomer, 2-acrylamidophenylboronic acid (2APBA), where the internal coordination helped to stabilize cross-links formed at acidic and neutral pH. Two different hydrogels were formed from a 2APBA copolymer cross-linked with either poly(vinyl alcohol) or a catechol-functionalized copolymer. The self-healing ability of these hydrogels was characterized through physical testing and rheological studies. Furthermore, the catechol cross-linked hydrogel was shown to be oxygen sensitive, demonstrating reduced self-healing and stress relaxation after partial oxidation. The synthesis of these hydrogels demonstrates a new strategy to produce boronic acid materials capable of self-healing at physiological pH.
Co-reporter:C. Adrian Figg, Tomohiro Kubo, and Brent S. Sumerlin
ACS Macro Letters 2015 Volume 4(Issue 10) pp:1114
Publication Date(Web):September 18, 2015
DOI:10.1021/acsmacrolett.5b00634
We report a strategy for the preparation of semitelechelic polymers containing two distinct functionalities at one chain end by consecutive and chemoselective nucleophilic aromatic substitution reactions on 2,4,6-trichloro-1,3,5-triazine (TCT). Because of its commercial availability, well-defined nature, and ubiquity in biological applications, monomethyl ether poly(ethylene glycol) (mPEG) was chosen to demonstrate the utility of this ω,ω-heterodifunctional end-group modification strategy. TCT-functionalized mPEG underwent highly efficient ω,ω-heterodisubstitution via sequential chemoselective substitution with model thiols and amines. The efficiency of nucleophile conjugation to the polymer end group was confirmed by 1H NMR spectroscopy and matrix assisted laser desorption-ionization time-of-flight mass spectrometry. In addition, density functional theory calculations provided insight into the importance of nucleophile addition order. This route introduces TCT derivatization as a powerful and facile tool to achieve specific polymeric end-group complexity and efficient heterogeneous functionalization.
Co-reporter:Jianbo Tan, Hao Sun, Mingguang Yu, Brent S. Sumerlin, and Li Zhang
ACS Macro Letters 2015 Volume 4(Issue 11) pp:1249
Publication Date(Web):October 26, 2015
DOI:10.1021/acsmacrolett.5b00748
Herein we report an aqueous photoinitiated polymerization-induced self-assembly (photo-PISA) for the preparation of a remarkably diverse set of complex polymer nanoparticle morphologies (e.g., spheres, worms, and vesicles) at room temperature. Ultrafast polymerization rates were achieved, with near quantitative monomer conversion within 15 min of visible light irradiation. An important feature of the photo-PISA is that diblock copolymer vesicles can be prepared under mild conditions (room temperature, aqueous medium, visible light), which will be important for the preparation of functional vesicles loaded with biorelated species (e.g., proteins). As a proof of concept, silica nanoparticles and bovine serum albumin (BSA) were encapsulated in situ within vesicles via the photo-PISA process.
Co-reporter:Bryan S. Tucker, Stephen G. Getchell, Megan R. Hill and Brent S. Sumerlin
Polymer Chemistry 2015 vol. 6(Issue 23) pp:4258-4263
Publication Date(Web):13 May 2015
DOI:10.1039/C5PY00497G
Poly(N-(2-hydroxypropyl)methacrylamide) (PHPMA), a biocompatible and non-immunogenic polymer, was used to form core-crosslinked star polymers for potential drug delivery applications. The conditions for the formation of the PHPMA stars were studied by varying the molecular weight of the PHPMA unimers, [crosslinker]:[unimer] ratios, and solvent. The optimized conditions were then used to form drug-loaded PHPMA star polymers by directly copolymerizing an HPMA-modified anticancer drug, methotrexate, during the crosslinking reaction of PHPMA unimers. The incorporation of the drug was confirmed by 1H NMR spectroscopy, and UV-visible spectroscopy was used to determine a drug loading of 20 wt%. Our initial drug release studies showed that the addition of an esterase induced drug release.
Co-reporter:Sunirmal Pal, Megan R. Hill and Brent S. Sumerlin
Polymer Chemistry 2015 vol. 6(Issue 45) pp:7871-7880
Publication Date(Web):28 Sep 2015
DOI:10.1039/C5PY01295C
Thermo- and redox-responsive hyperbranched copolymers were prepared by statistical copolymerization of N-isopropylacrylamide (NIPAM) and N,N′-bis(acryloyl)cystamine (BAC) by reversible addition–fragmentation chain transfer (RAFT) polymerization. Kinetic studies revealed that the molecular weight of the resulting poly(NIPAM-co-BAC) gradually increased during the polymerization, and control over molecular weight and degree of branching was demonstrated. The hyperbranched copolymers showed thermoresponsive self-assembly, as determined by dynamic light scattering and turbidity measurements. Hyperbranched poly(NIPAM-co-BAC) copolymers were further used for the synthesis of star copolymers by chain extension with N,N-dimethylacrylamide. The resulting star copolymers with hyperbranched cores and linear arms were readily degraded under reducing conditions due to the divinyl crosslinker containing a redox-sensitive disulfide linkage. Not only did the disulfide bonds result in macromolecules with redox-responsive behavior, but the ability to cleave and characterize the individual branches of the hyperbranch polymer after synthesis allowed us to confirm the controlled nature of RAFT polymerization in the presence of divinyl compounds.
Co-reporter:Patricia R. Bachler;Michael D. Schulz;Chelsea A. Sparks;Kenneth B. Wagener
Macromolecular Rapid Communications 2015 Volume 36( Issue 9) pp:828-833
Publication Date(Web):
DOI:10.1002/marc.201500060
Co-reporter:Soma Mukherjee, Megan R. Hill and Brent S. Sumerlin
Soft Matter 2015 vol. 11(Issue 30) pp:6152-6161
Publication Date(Web):06 Jul 2015
DOI:10.1039/C5SM00865D
Self-healing oxime-functional hydrogels have been developed that undergo a reversible gel-to-sol transition via oxime exchange under acidic conditions. Keto-functional copolymers were prepared by conventional radical polymerization of N,N-dimethylacrylamide (DMA) and diacetone acrylamide (DAA). The resulting water soluble copolymers (P(DMA-stat-DAA)) were chemically crosslinked with difunctional alkoxyamines to obtain hydrogels via oxime formation. Gel-to-sol transitions were induced by the addition of excess monofunctional alkoxyamines to promote competitive oxime exchange under acidic conditions at 25 °C. The hydrogel could autonomously heal after it was damaged due to the dynamic nature of the oxime crosslinks. In addition to their chemo-responsive behavior, the P(DMA-stat-DAA) copolymers exhibit cloud points which vary with the DAA content in the copolymers. This thermo-responsive behavior of the P(DMA-stat-DAA) was utilized to form physical hydrogels above their cloud point. Therefore, these materials can either form dynamic-covalent or physically-crosslinked gels, both of which demonstrate reversible gelation behavior.
Co-reporter:Jessica J. Cash, Tomohiro Kubo, Abhijeet P. Bapat, and Brent S. Sumerlin
Macromolecules 2015 Volume 48(Issue 7) pp:2098-2106
Publication Date(Web):March 26, 2015
DOI:10.1021/acs.macromol.5b00210
Cross-linked polymers constructed with dynamic-covalent boronic esters were synthesized via photoinitiated radical thiol–ene click chemistry. Because the reversibility of the boronic ester cross-links was readily accessible, the resulting materials were capable of undergoing bond exchange to covalently mend after failure. The reversible bonds of the boronic esters were shown to shift their exchange equilibrium at room temperature when exposed to water. Nevertheless, the materials were observed to be stable and hydrophobic and absorbed only minor amounts of water over extended periods of time when submerged in water or exposed to humid environments. The facile reversibility of the networks allowed intrinsic self-healing under ambient conditions. Highly efficient self-healing of these bulk materials was confirmed by mechanical testing, even after subjecting a single site to multiple cut–repair cycles. Several variables were considered for their effect on materials properties and healing, including cross-link density, humidity, and healing time.
Co-reporter:Megan R. Hill, R. Nicholas Carmean, and Brent S. Sumerlin
Macromolecules 2015 Volume 48(Issue 16) pp:5459-5469
Publication Date(Web):July 28, 2015
DOI:10.1021/acs.macromol.5b00342
Reversible deactivation radical polymerization (RDRP) has revolutionized modern polymer chemistry over the past two decades, thus laying the groundwork for the synthesis of complex macromolecules and enabling the preparation of previously inaccessible materials. Reversible addition-fragmentation chain transfer (RAFT) polymerization has emerged as one of the most promising techniques because of its functional group tolerance, applicability to a wide range of vinyl monomers, and its nondemanding experimental conditions. However, despite the promise and clearly demonstrated utility of RAFT, limitations of the method sometimes still exist, including the occasional need for extended polymerization times, limited access to high molecular weight polymers, low “livingness” due to unavoidable radical termination events, etc. This Perspective focuses on recent advances that have been specifically designed to address many of these perceived limitations to reinforce the promise of RAFT for the synthesis of complex and well-defined polymers under facile conditions.
Co-reporter:Megan R. Hill, Elliot J. MacKrell, Carl P. Forsthoefel, Shaun P. Jensen, Mingsheng Chen, Gloria A. Moore, Zhenli L. He, and Brent S. Sumerlin
Biomacromolecules 2015 Volume 16(Issue 4) pp:
Publication Date(Web):March 10, 2015
DOI:10.1021/acs.biomac.5b00069
We report the synthesis and characterization of pH-responsive polysuccinimide-based nanoparticles. Polysuccinimide (PSI), a precursor to biodegradable poly(aspartic acid), was synthesized from the condensation of l-aspartic acid and subsequently functionalized with primary amines to form random amphiphilic copolymers. The copolymers formed stable nanoparticles in aqueous medium via nanoprecipitation and were subsequently loaded with a model hydrophobic molecule to demonstrate their potential as controlled-release delivery vehicles. It was found that above pH 7, the hydrophobic succinimidyl units of the PSI nanoparticles hydrolyzed to release encapsulated materials. The release rate significantly increased at elevated pH and decreased with an increasing degree of functionalization. Finally, plant toxicity studies showed that the polymer materials exhibit little to no toxic effects at biologically relevant concentrations.
Co-reporter:Bryan S. Tucker, Jon D. Stewart, J. Ignacio Aguirre, L. Shannon Holliday, C. Adrian Figg, Jonathan G. Messer, and Brent S. Sumerlin
Biomacromolecules 2015 Volume 16(Issue 8) pp:
Publication Date(Web):July 7, 2015
DOI:10.1021/acs.biomac.5b00623
Polymers of similar molecular weights and chemical constitution but varying in their macromolecular architectures were conjugated to osteoprotegerin (OPG) to determine the effect of polymer topology on protein activity in vitro and in vivo. OPG is a protein that inhibits bone resorption by preventing the formation of mature osteoclasts from the osteoclast precursor cell. Accelerated bone loss disorders, such as osteoporosis, rheumatoid arthritis, and metastatic bone disease, occur as a result of increased osteoclastogenesis, leading to the severe weakening of the bone. OPG has shown promise as a treatment in bone disorders; however, it is rapidly cleared from circulation through rapid liver uptake, and frequent, high doses of the protein are necessary to achieve a therapeutic benefit. We aimed to improve the effectiveness of OPG by creating OPG–polymer bioconjugates, employing reversible addition–fragmentation chain transfer polymerization to create well-defined polymers with branching densities varying from linear, loosely branched to densely branched. Polymers with each of these architectures were conjugated to OPG using a “grafting-to” approach, and the bioconjugates were characterized by sodium dodecyl sulfate polyacrylamide gel electrophoresis. The OPG–polymer bioconjugates showed retention of activity in vitro against osteoclasts, and each bioconjugate was shown to be nontoxic. Preliminary in vivo studies further supported the nontoxic characteristics of the bioconjugates, and measurement of the bone mineral density in rats 7 days post-treatment via peripheral quantitative computed tomography suggested a slight increase in bone mineral density after administration of the loosely branched OPG–polymer bioconjugate.
Co-reporter:Hao Sun, Christopher P. Kabb and Brent S. Sumerlin
Chemical Science 2014 vol. 5(Issue 12) pp:4646-4655
Publication Date(Web):2014/08/28
DOI:10.1039/C4SC02290D
A thermally-reversible inimer was used to confirm the controlled growth of individual branches during self-condensing vinyl atom transfer radical polymerization (ATRP). Segmented hyperbranched polymers were synthesized by ATRP of methyl methacrylate (MMA) and a novel inimer that contained a thermally labile Diels–Alder linkage between its initiating and polymerizable moieties. Three distinct feed ratios of MMA to inimer (15:1, 30:1, and 60:1) yielded hyperbranched polymers with variable degrees of branching and molecular weights in the range of 120000 to 515000 g mol−1. The resulting hyperbranched polymers contained thermally-reversible branch points that were cleaved quantitatively on heating to yield linear polymers with molecular weights that were similar to the theoretical values that would be expected based on controlled chain growth of individual branches during self-condensing vinyl polymerization (SCVP). The cleaved linear polymers contained pendant furan and terminal maleimide functionalities that allowed reassembly at 50 °C to form “healed” hyperbranched polymers. The healing efficiency was determined by 1H NMR spectroscopy, and the molecular weights of the repaired hyperbranched polymers were characterized by gel permeation chromatography. A segmented hyperbranched polymer was employed as a multifunctional macroinitiator to prepare an amphiphilic “hyper-star” via chain extension with poly(ethylene glycol) methyl ether methacrylate. Assembly of these “hyper-stars” into well-defined micelles (∼23 nm) in neutral water was confirmed by transmission electron microscopy and dynamic light scattering.
Co-reporter:Fransiska S. H. Krismastuti, William L. A. Brooks, Martin J. Sweetman, Brent S. Sumerlin and Nicolas H. Voelcker
Journal of Materials Chemistry A 2014 vol. 2(Issue 25) pp:3972-3983
Publication Date(Web):07 May 2014
DOI:10.1039/C4TB00231H
The ability to monitor glucose levels in chronic wound fluid of diabetic patients is a promising theranostic approach in chronic wound healing. Phenylboronic acid polymers are glucose- and pH-responsive materials. In the presence of glucose, these polymers reversibly form cyclic boronate esters, changing the properties of the polymer and forming the basis of glucose sensing. In this report, poly(4-vinylphenylboronic acid) (PVPBA) was covalently grafted to the pores of porous silicon (pSi) films (pSi-PVPBA). Polymer switching in response to changing pH and glucose concentration was monitored by means of interferometric reflectance spectroscopy (IRS). We observed that a shift of the boronic acid equilibrium between the neutral and anionic form in the polymer translated into refractive index changes that could be detected as a variation of the effective optical thickness (EOT) of the pSi-PVPBA film. The pSi/polymer composite was further investigated as a platform for the detection of glucose. Using this sensing platform, we were able to detect glucose in a buffer solution as low as 0.15 mM and also in a wound fluid sample without encountering interferences.
Co-reporter:Bryan S. Tucker and Brent S. Sumerlin
Polymer Chemistry 2014 vol. 5(Issue 5) pp:1566-1572
Publication Date(Web):11 Oct 2013
DOI:10.1039/C3PY01279D
This mini-review provides a brief overview of recent advances in the area of biologically-relevant nanomaterials composed of poly(N-(2-hydroxypropyl) methacrylamide) (PHPMA). Polymer diversity within the field of nanomedicine has grown considerably in recent years, yet the overwhelming majority of soft nanomaterials intended for medical applications are composed of poly(ethylene glycol) (PEG) and its derivatives. However, it is well known that PHPMA offers several advantages over PEG in some applications, including its ability to be functionalized via its side-chain hydroxyl group to incorporate drugs, imaging agents, targeting ligands, etc. This mini-review focuses on select recent advances in PHPMA-based nanostructures. Particular attention is placed on polymer–drug conjugates, self-assembled nanoparticles, and other recent examples of PHPMA-based nanotherapeutics.
Co-reporter:Soma Mukherjee, Abhijeet P. Bapat, Megan R. Hill and Brent S. Sumerlin
Polymer Chemistry 2014 vol. 5(Issue 24) pp:6923-6931
Publication Date(Web):09 Oct 2014
DOI:10.1039/C4PY01282H
We demonstrate the formation of oxime-functional macromolecular stars that are able to dissociate and reconstruct themselves upon application of a stimulus. The reversible nature of the oxime bond in the presence of externally added alkoxyamines or carbonyl compounds enables reconfiguration via competitive exchange. Reversible addition–fragmentation chain transfer (RAFT) polymerization was utilized to prepare well-defined amphiphilic block copolymers in which a hydrophobic keto-functional block allowed self-assembly into micelles in water. Adding a difunctional alkoxyamine small molecule to these solutions resulted in crosslinking of the micelles to yield macromolecular stars. The reversible nature of the O-alkyl oxime linkages was demonstrated via competitive exchange with excess of carbonyl compounds or monofunctional alkoxyamine under acidic conditions and at elevated temperatures to result in dissociation of the stars to unimolecular oxime-functional polymer chains.
Co-reporter:Debashish Roy
Macromolecular Rapid Communications 2014 Volume 35( Issue 2) pp:174-179
Publication Date(Web):
DOI:10.1002/marc.201300642
Co-reporter:Debashish Roy, William L. A. Brooks and Brent S. Sumerlin
Chemical Society Reviews 2013 vol. 42(Issue 17) pp:7214-7243
Publication Date(Web):28 Feb 2013
DOI:10.1039/C3CS35499G
Interest in thermoresponsive polymers has steadily grown over many decades, and a great deal of work has been dedicated to developing temperature sensitive macromolecules that can be crafted into new smart materials. However, the overwhelming majority of previously reported temperature-responsive polymers are based on poly(N-isopropylacrylamide) (PNIPAM), despite the fact that a wide range of other thermoresponsive polymers have demonstrated similar promise for the preparation of adaptive materials. Herein, we aim to highlight recent results that involve thermoresponsive systems that have not yet been as fully considered. Many of these (co)polymers represent clear opportunities for advancements in emerging biomedical and materials fields due to their increased biocompatibility and tuneable response. By highlighting recent examples of newly developed thermoresponsive polymer systems, we hope to promote the development of new generations of smart materials.
Co-reporter:Priyadarsi De
Macromolecular Chemistry and Physics 2013 Volume 214( Issue 2) pp:272-279
Publication Date(Web):
DOI:10.1002/macp.201200416
Abstract
The readily tuned thermoresponsive behavior of well-defined statistical and block copolymers of di(ethylene glycol)ethyl ether acrylate (DEGA) and N,N-dimethylacrylamide (DMA) was investigated. DEGA and DMA were copolymerized with various comonomer feed ratios by reversible addition–fragmentation chain transfer (RAFT) polymerization. Reactivity ratios were determined, which allowed copolymers with precisely tuned compositions to be prepared. The cloud point of the resulting copolymers was observed to vary linearly with the mole fraction of hydrophilic DMA over the range of 10–90 °C. Block copolymers of DEGA were also prepared by chain extension of poly(DEGA) homopolymers with DMA. The resulting block copolymers formed well-defined polymeric micelles when heated above the cloud point of the poly(DEGA) block. Thus, copolymerization of DEGA was shown to be a convenient route to thermoresponsive polymers with specific and predictable transition temperatures.
Co-reporter:Abhijeet P. Bapat, Jacob G. Ray, Daniel A. Savin, and Brent S. Sumerlin
Macromolecules 2013 Volume 46(Issue 6) pp:
Publication Date(Web):March 8, 2013
DOI:10.1021/ma400169m
Dynamic-covalent macromolecular stars were prepared by cross-linking block copolymers containing reactive maleic anhydride units with a disulfide-containing diamine. Here we report the synthesis of disulfide-cross-linked star polymers obtained by the arm-first process. Well-defined block copolymers containing a reactive poly(styrene-alt-maleic anhydride) (P(S-alt-MAn)) segment and an inert polystyrene or poly(N-isopropylacrylamide) segment were obtained by reversible addition–fragmentation chain transfer (RAFT) polymerization. Facile ring-opening of the pendant anhydride groups in the block copolymers by a disulfide-linked diamine cross-linker led to core-cross-linked stars with redox-responsive cores. The reductive cleavage of the disulfide linkages in the cross-linked cores resulted in star dissociation into linear arms with pendant thiol groups. Oxidation of the pendant thiol units of the resulting unimers in the presence of air led to reassembly or self-healing of the stars without the need for an externally added oxidizing agent.
Co-reporter:Hao Sun, Christopher P. Kabb and Brent S. Sumerlin
Chemical Science (2010-Present) 2014 - vol. 5(Issue 12) pp:NaN4655-4655
Publication Date(Web):2014/08/28
DOI:10.1039/C4SC02290D
A thermally-reversible inimer was used to confirm the controlled growth of individual branches during self-condensing vinyl atom transfer radical polymerization (ATRP). Segmented hyperbranched polymers were synthesized by ATRP of methyl methacrylate (MMA) and a novel inimer that contained a thermally labile Diels–Alder linkage between its initiating and polymerizable moieties. Three distinct feed ratios of MMA to inimer (15:1, 30:1, and 60:1) yielded hyperbranched polymers with variable degrees of branching and molecular weights in the range of 120000 to 515000 g mol−1. The resulting hyperbranched polymers contained thermally-reversible branch points that were cleaved quantitatively on heating to yield linear polymers with molecular weights that were similar to the theoretical values that would be expected based on controlled chain growth of individual branches during self-condensing vinyl polymerization (SCVP). The cleaved linear polymers contained pendant furan and terminal maleimide functionalities that allowed reassembly at 50 °C to form “healed” hyperbranched polymers. The healing efficiency was determined by 1H NMR spectroscopy, and the molecular weights of the repaired hyperbranched polymers were characterized by gel permeation chromatography. A segmented hyperbranched polymer was employed as a multifunctional macroinitiator to prepare an amphiphilic “hyper-star” via chain extension with poly(ethylene glycol) methyl ether methacrylate. Assembly of these “hyper-stars” into well-defined micelles (∼23 nm) in neutral water was confirmed by transmission electron microscopy and dynamic light scattering.
Co-reporter:Fransiska S. H. Krismastuti, William L. A. Brooks, Martin J. Sweetman, Brent S. Sumerlin and Nicolas H. Voelcker
Journal of Materials Chemistry A 2014 - vol. 2(Issue 25) pp:NaN3983-3983
Publication Date(Web):2014/05/07
DOI:10.1039/C4TB00231H
The ability to monitor glucose levels in chronic wound fluid of diabetic patients is a promising theranostic approach in chronic wound healing. Phenylboronic acid polymers are glucose- and pH-responsive materials. In the presence of glucose, these polymers reversibly form cyclic boronate esters, changing the properties of the polymer and forming the basis of glucose sensing. In this report, poly(4-vinylphenylboronic acid) (PVPBA) was covalently grafted to the pores of porous silicon (pSi) films (pSi-PVPBA). Polymer switching in response to changing pH and glucose concentration was monitored by means of interferometric reflectance spectroscopy (IRS). We observed that a shift of the boronic acid equilibrium between the neutral and anionic form in the polymer translated into refractive index changes that could be detected as a variation of the effective optical thickness (EOT) of the pSi-PVPBA film. The pSi/polymer composite was further investigated as a platform for the detection of glucose. Using this sensing platform, we were able to detect glucose in a buffer solution as low as 0.15 mM and also in a wound fluid sample without encountering interferences.
Co-reporter:Debashish Roy, William L. A. Brooks and Brent S. Sumerlin
Chemical Society Reviews 2013 - vol. 42(Issue 17) pp:NaN7243-7243
Publication Date(Web):2013/02/28
DOI:10.1039/C3CS35499G
Interest in thermoresponsive polymers has steadily grown over many decades, and a great deal of work has been dedicated to developing temperature sensitive macromolecules that can be crafted into new smart materials. However, the overwhelming majority of previously reported temperature-responsive polymers are based on poly(N-isopropylacrylamide) (PNIPAM), despite the fact that a wide range of other thermoresponsive polymers have demonstrated similar promise for the preparation of adaptive materials. Herein, we aim to highlight recent results that involve thermoresponsive systems that have not yet been as fully considered. Many of these (co)polymers represent clear opportunities for advancements in emerging biomedical and materials fields due to their increased biocompatibility and tuneable response. By highlighting recent examples of newly developed thermoresponsive polymer systems, we hope to promote the development of new generations of smart materials.
Co-reporter:Yuqiong Dai, Hao Sun, Sunirmal Pal, Yunlu Zhang, Sangwoo Park, Christopher P. Kabb, Wei David Wei and Brent S. Sumerlin
Chemical Science (2010-Present) 2017 - vol. 8(Issue 3) pp:NaN1821-1821
Publication Date(Web):2016/12/13
DOI:10.1039/C6SC04650A
Responsive systems sensitive to near-infrared (NIR) light are promising for triggered release due to efficient deep tissue penetration of NIR irradiation relative to higher energy sources (e.g., UV), allowing for spatiotemporal control over triggering events with minimal potential for tissue damage. Herein, we report star polymers containing thermally-labile azo linkages that dissociate during conventional heating or during localized heating via the photothermal effect upon NIR irradiation. Controlled release during conventional heating was investigated for the star polymers loaded with a model dye, with negligible release being observed at 25 °C and >80% release at 90 °C. Star polymers co-loaded with NIR-responsive indocyanine green showed rapid dye release upon NIR irradiation (λ ≥ 715 nm) due to the photothermally-induced degradation of azo linkages within the cores of the star polymers. This approach provides access to a new class of delivery and release systems that can be triggered by noninvasive external stimulation.
Co-reporter:Mingsheng Chen, Shaun P. Jensen, Megan R. Hill, Gloria Moore, Zhenli He and Brent S. Sumerlin
Chemical Communications 2015 - vol. 51(Issue 47) pp:NaN9697-9697
Publication Date(Web):2015/05/15
DOI:10.1039/C5CC02726H
While polymeric nanocarriers are widely used in medicine for controlled release and site-specific delivery, few reports have applied such delivery methods within agriculture, despite the urgent need for specific delivery of pesticides and nutrients. We report the synthesis of stimuli-responsive and biodegradable polymeric nanocarriers designed for delivery to the phloem of plants and describe methods employed to evaluate their toxicity in plant cells.
Co-reporter:Christopher P. Kabb, R. Nicholas Carmean and Brent S. Sumerlin
Chemical Science (2010-Present) 2015 - vol. 6(Issue 10) pp:NaN5669-5669
Publication Date(Web):2015/07/10
DOI:10.1039/C5SC01535A
The surface-localized hyperthermia of gold nanoparticles under microwave irradiation was examined. Gold nanoparticles with a hydrodynamic diameter of ∼6 nm stabilized by polymeric “thermometers” were used to gather information on the extent of heating as well as its spatial confinements. Reversible addition–fragmentation chain transfer polymerization was employed to synthesize well-defined, functional polymers of predetermined molecular weights, allowing for estimation of the distance between the nanoparticle surface and the polymer chain end. The polymers were conjugated with a fluorescent dye separated by a thermally-labile azo linkage, and these polymeric ligands were bound to gold nanoparticles via gold–thiolate bonds. Conventional heating experiments elucidated the relationship between temperature and the extent of dye release from the gold nanoparticle using fluorescence spectroscopy. The local temperature increase experienced under microwave irradiation was calculated using the same methodology. This approach indicated the temperature near the surface of the nanoparticle was nearly 70 °C higher than the bulk solution temperature, but decreased rapidly with distance, with no noticeable temperature increase when the azo linkage was approximately 2 nm away.
Co-reporter:C. Adrian Figg, Alexandre Simula, Kalkidan A. Gebre, Bryan S. Tucker, David M. Haddleton and Brent S. Sumerlin
Chemical Science (2010-Present) 2015 - vol. 6(Issue 2) pp:NaN1236-1236
Publication Date(Web):2014/11/14
DOI:10.1039/C4SC03334E
Polymerization-induced self-assembly (PISA) is a versatile technique to achieve a wide range of polymeric nanoparticle morphologies. Most previous examples of self-assembled soft nanoparticle synthesis by PISA rely on a growing solvophobic polymer block that leads to changes in nanoparticle architecture during polymerization in a selective solvent. However, synthesis of block copolymers with a growing stimuli-responsive block to form various nanoparticle shapes has yet to be reported. This new concept using thermoresponsive polymers is termed polymerization-induced thermal self-assembly (PITSA). A reversible addition-fragmentation chain transfer (RAFT) polymerization of N-isopropylacrylamide from a hydrophilic chain transfer agent composed of N,N-dimethylacrylamide and acrylic acid was carried out in water above the known lower critical solution temperature (LCST) of poly(N-isopropylacrylamide) (PNIPAm). After reaching a certain chain length, the growing PNIPAm self-assembled, as induced by the LCST, into block copolymer aggregates within which dispersion polymerization continued. To characterize the nanoparticles at ambient temperatures without their dissolution, the particles were crosslinked immediately following polymerization at elevated temperatures via the reaction of the acid groups with a diamine in the presence of a carbodiimide. Size exclusion chromatography was used to evaluate the unimer molecular weight distributions and reaction kinetics. Dynamic light scattering and transmission electron microscopy provided insight into the size and morphologies of the nanoparticles. The resulting block copolymers formed polymeric nanoparticles with a range of morphologies (e.g., micelles, worms, and vesicles), which were a function of the PNIPAm block length.


![Propanoic acid, 2-[[[(2-carboxyethyl)thio]thioxomethyl]thio]-2-methyl-](/data/chemimg/3502700/870451-10-8.png)
![Propanoic acid, 2-[[[(2-carboxyethyl)thio]thioxomethyl]thio]-2-methyl-](/data/chemimg/3502700/870451-10-8_b.png)

















![Poly[1,3-cyclopentanediyl-(1Z)-1,2-ethenediyl]](http://img.cochemist.com/ccimg/65600/65556-06-1.png)
![Poly[1,3-cyclopentanediyl-(1Z)-1,2-ethenediyl]](http://img.cochemist.com/ccimg/65600/65556-06-1_b.png)
![(4-[(9H-FLUOREN-9-YLMETHOXY)CARBONYL]-1-{[(2-METHYL-2-PROPANYL)OX<WBR />Y]CARBONYL}-2-PIPERAZINYL)ACETIC ACID](http://img.cochemist.com/ccimg/34100/34063-67-7.png)
![(4-[(9H-FLUOREN-9-YLMETHOXY)CARBONYL]-1-{[(2-METHYL-2-PROPANYL)OX<WBR />Y]CARBONYL}-2-PIPERAZINYL)ACETIC ACID](http://img.cochemist.com/ccimg/34100/34063-67-7_b.png)
![Phosphonic acid, P,P'-[[(2-hydroxyethyl)imino]bis(methylene)]bis-, P,P,P',P'-tetraethyl ester](/data/chemimg/504400/29920-33-0.png)
![Phosphonic acid, P,P'-[[(2-hydroxyethyl)imino]bis(methylene)]bis-, P,P,P',P'-tetraethyl ester](/data/chemimg/504400/29920-33-0_b.png)
![Poly[imino[(1S)-1-(carboxymethyl)-2-oxo-1,2-ethanediyl]]](http://img.cochemist.com/ccimg/26100/26063-13-8.png)
![Poly[imino[(1S)-1-(carboxymethyl)-2-oxo-1,2-ethanediyl]]](http://img.cochemist.com/ccimg/26100/26063-13-8_b.png)
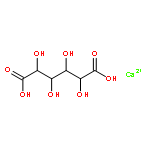
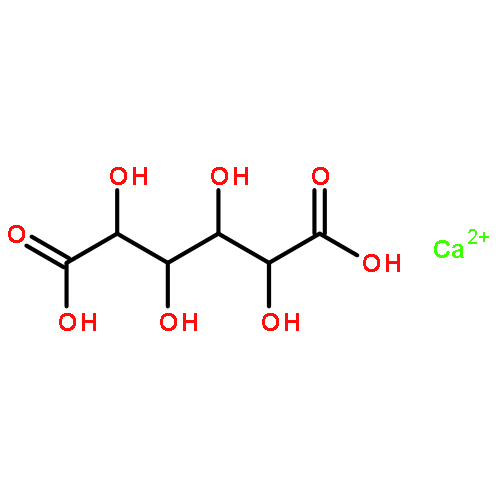
![1-[6-(2,5-dioxopyrrol-1-yl)hexyl]pyrrole-2,5-dione](http://img.cochemist.com/ccimg/4900/4856-87-5.png)
![1-[6-(2,5-dioxopyrrol-1-yl)hexyl]pyrrole-2,5-dione](http://img.cochemist.com/ccimg/4900/4856-87-5_b.png)
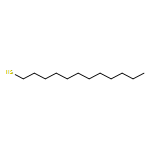
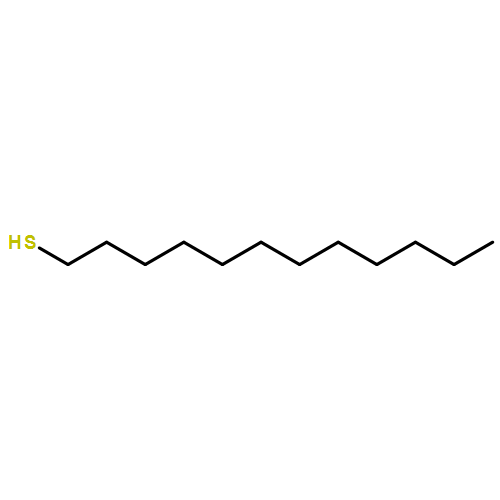









![Propanoic acid, 2-[[(ethylthio)thioxomethyl]thio]-2-methyl-](/data/chemimg/113500/881037-62-3.png)
![Propanoic acid, 2-[[(ethylthio)thioxomethyl]thio]-2-methyl-](/data/chemimg/113500/881037-62-3_b.png)
![Carbamic acid, [2-[(1-oxo-2-propenyl)amino]ethyl]-, 1,1-dimethylethylester](http://img.cochemist.com/ccimg/165200/165196-44-1.png)
![Carbamic acid, [2-[(1-oxo-2-propenyl)amino]ethyl]-, 1,1-dimethylethylester](http://img.cochemist.com/ccimg/165200/165196-44-1_b.png)



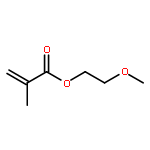
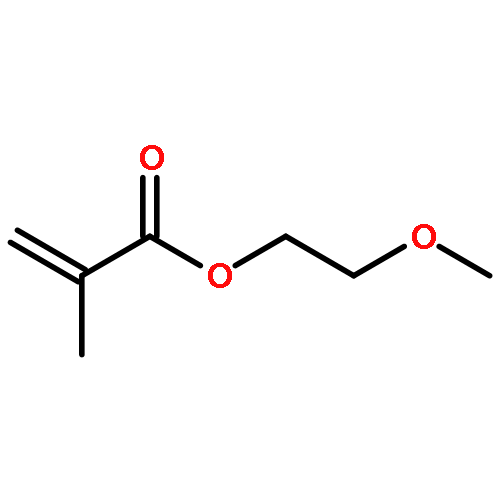
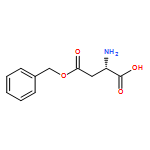
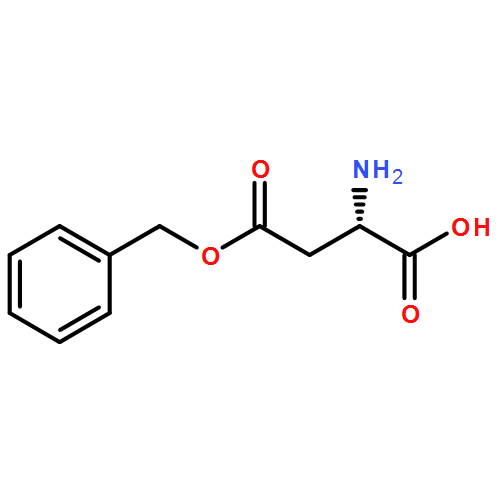
![5H-Benzo[a]phenoxazin-5-one,9-(diethylamino)-](http://img.cochemist.com/ccimg/7400/7385-67-3.png)
![5H-Benzo[a]phenoxazin-5-one,9-(diethylamino)-](http://img.cochemist.com/ccimg/7400/7385-67-3_b.png)