Co-reporter:Kelli Kazmier, Derek P Claxton, Hassane S Mchaourab
Current Opinion in Structural Biology 2017 Volume 45(Volume 45) pp:
Publication Date(Web):1 August 2017
DOI:10.1016/j.sbi.2016.12.006
•Alternating access in the LeuT-fold class of transporters is discussed.•Structural analysis suggests mechanistic divergence among class members.•Ligand-dependent conformational equilibria is a defining feature of transport.•Descriptions of transport energy landscapes require a multifaceted approach.Secondary active transporters couple the uphill translocation of substrates to electrochemical ion gradients. Transporter conformational motion, generically referred to as alternating access, enables a central ligand binding site to change its orientation relative to the membrane. Here we review themes of alternating access and the transduction of ion gradient energy to power this process in the LeuT-fold class of transporters where crystallographic, computational and spectroscopic approaches have converged to yield detailed models of transport cycles. Specifically, we compare findings for the Na+-coupled amino acid transporter LeuT and the Na+-coupled hydantoin transporter Mhp1. Although these studies have illuminated multiple aspects of transporter structures and dynamics, a number of questions remain unresolved that so far hinder understanding transport mechanisms in an energy landscape perspective.Download high-res image (157KB)Download full-size image
Co-reporter:Alberto Collauto, Smriti Mishra, Aleksei Litvinov, Hassane S. Mchaourab, Daniella Goldfarb
Structure 2017 Volume 25, Issue 8(Volume 25, Issue 8) pp:
Publication Date(Web):1 August 2017
DOI:10.1016/j.str.2017.06.014
•Detection of ATP hydrolysis by hyperfine selective EPR of Mn2+•Different coordination of ATP by MsbA and BmrCD•Detection of vanadate reveals asymmetric hydrolysis of ATPWe have applied high-field (W-band) pulse electron-nuclear double resonance (ENDOR) and electron-electron double resonance (ELDOR)-detected nuclear magnetic resonance (EDNMR) to characterize the coordination sphere of the Mn2+ co-factor in the nucleotide binding sites (NBSs) of ABC transporters. MsbA and BmrCD are two efflux transporters hypothesized to represent divergent catalytic mechanisms. Our results reveal distinct coordination of Mn2+ to ATP and transporter residues in the consensus and degenerate NBSs of BmrCD. In contrast, the coordination of Mn2+ at the two NBSs of MsbA is similar, which provides a mechanistic rationale for its higher rate constant of ATP hydrolysis relative to BmrCD. Direct detection of vanadate ion, trapped in a high-energy post-hydrolysis intermediate, further supports the notion of asymmetric hydrolysis by the two NBSs of BmrCD. The integrated spectroscopic approach presented here, which link energy input to conformational dynamics, can be applied to a variety of systems powered by ATP turnover.Download high-res image (170KB)Download full-size image
Co-reporter:Reza Dastvan;Axel W. Fischer;Smriti Mishra;Jens Meiler
PNAS 2016 Volume 113 (Issue 5 ) pp:1220-1225
Publication Date(Web):2016-02-02
DOI:10.1073/pnas.1520431113
The small multidrug transporter from Escherichia coli, EmrE, couples the energetically uphill extrusion of hydrophobic cations out of the cell to the transport of two protons
down their electrochemical gradient. Although principal mechanistic elements of proton/substrate antiport have been described,
the structural record is limited to the conformation of the substrate-bound state, which has been shown to undergo isoenergetic
alternating access. A central but missing link in the structure/mechanism relationship is a description of the proton-bound
state, which is an obligatory intermediate in the transport cycle. Here we report a systematic spin labeling and double electron
electron resonance (DEER) study that uncovers the conformational changes of EmrE subsequent to protonation of critical acidic
residues in the context of a global description of ligand-induced structural rearrangements. We find that protonation of E14
leads to extensive rotation and tilt of transmembrane helices 1–3 in conjunction with repacking of loops, conformational changes
that alter the coordination of the bound substrate and modulate its access to the binding site from the lipid bilayer. The
transport model that emerges from our data posits a proton-bound, but occluded, resting state. Substrate binding from the
inner leaflet of the bilayer releases the protons and triggers alternating access between inward- and outward-facing conformations
of the substrate-loaded transporter, thus enabling antiport without dissipation of the proton gradient.
Co-reporter:Hanane A. Koteiche, Derek P. Claxton, Sanjay Mishra, Richard A. Stein, Ezelle T. McDonald, and Hassane S. Mchaourab
Biochemistry 2015 Volume 54(Issue 38) pp:
Publication Date(Web):September 17, 2015
DOI:10.1021/acs.biochem.5b00678
In addition to contributing to lens optical properties, the α-crystallins are small heat shock proteins that possess chaperone activity and are predicted to bind and sequester destabilized proteins to delay cataract formation. The current model of α-crystallin chaperone mechanism envisions a transition from the native oligomer to an activated form that has higher affinity to non-native states of the substrate. Previous studies have suggested that this oligomeric plasticity is encoded in the primary sequence and controls access to high affinity binding sites within the N-terminal domain. Here, we further examined the role of sequence variation in the context of species-specific α-crystallins from rat and zebrafish. Alternative splicing of the αA gene in rodents produces αAins, which is distinguished by a longer N-terminal domain. The zebrafish genome includes duplicate αB-crystallin genes, αBa and αBb, which display divergent primary sequence and tissue expression patterns. Equilibrium binding experiments were employed to quantitatively define chaperone interactions with a destabilized model substrate, T4 lysozyme. In combination with multiangle light scattering, we show that rat αAins and zebrafish α-crystallins display distinct global structural properties and chaperone activities. Notably, we find that αAins and αBa demonstrate substantially enhanced chaperone function relative to other α-crystallins, binding the same substrate more than 2 orders of magnitude higher affinity and mimicking the activity of fully activated mammalian small heat shock proteins. These results emphasize the role of sequence divergence as an evolutionary strategy to tune chaperone function to the requirements of the tissues and organisms in which they are expressed.
Co-reporter:Shahidul M. Islam;Shruti Sharma;Kelli Kazmier;Benoît Roux
PNAS 2014 Volume 111 (Issue 41 ) pp:14752-14757
Publication Date(Web):2014-10-14
DOI:10.1073/pnas.1410431111
Ion-dependent transporters of the LeuT-fold couple the uptake of physiologically essential molecules to transmembrane ion
gradients. Defined by a conserved 5-helix inverted repeat that encodes common principles of ion and substrate binding, the
LeuT-fold has been captured in outward-facing, occluded, and inward-facing conformations. However, fundamental questions relating
to the structural basis of alternating access and coupling to ion gradients remain unanswered. Here, we used distance measurements
between pairs of spin labels to define the conformational cycle of the Na+-coupled hydantoin symporter Mhp1 from Microbacterium liquefaciens. Our results reveal that the inward-facing and outward-facing Mhp1 crystal structures represent sampled intermediate states
in solution. Here, we provide a mechanistic context for these structures, mapping them into a model of transport based on
ion- and substrate-dependent conformational equilibria. In contrast to the Na+/leucine transporter LeuT, our results suggest that Na+ binding at the conserved second Na+ binding site does not change the energetics of the inward- and outward-facing conformations of Mhp1. Comparative analysis
of ligand-dependent alternating access in LeuT and Mhp1 lead us to propose that different coupling schemes to ion gradients
may define distinct conformational mechanisms within the LeuT-fold class.
Co-reporter:P. Ryan Steed, Richard A. Stein, Smriti Mishra, Michael C. Goodman, and Hassane S. Mchaourab
Biochemistry 2013 Volume 52(Issue 34) pp:
Publication Date(Web):July 31, 2013
DOI:10.1021/bi4008935
NorM of the multidrug and toxic compound extrusion (MATE) family of transporters couples the efflux of a broad range of hydrophobic molecules to an inward Na+ gradient across the cell membrane. Several crystal structures of MATE transporters revealed distinct substrate binding sites leading to differing models of the mechanism of ion-coupled substrate extrusion. In the experiments reported here, we observed that a spin-labeled derivative of daunorubicin, Ruboxyl, is transported by NorM from Vibrio cholerae. It is therefore ideal for characterizing mechanistically relevant binding interactions with NorM and directly addressing the coupling of ion and drug binding. Fluorescence and electron paramagnetic resonance experiments revealed that Ruboxyl binds to NorM with micromolar affinity and becomes immobilized upon binding, even in the presence of Na+. Using double electron–electron resonance spectroscopy, we determined that Ruboxyl binds to a single site on the periplasmic side of the protein. The presence of Na+ did not translocate the substrate to a second site as previously proposed. These experiments surprisingly show that Na+ does not affect the affinity or location of the substrate binding site on detergent-solubilized NorM, thus suggesting that additional factors beyond simple mutual exclusivity of binding, such as the presence of a Na+ gradient across the native membrane, govern Na+–drug coupling during antiport.
Co-reporter:P. Ryan Steed, Ping Zou, Kristin E. Trone, and Hassane S. Mchaourab
Biochemistry 2013 Volume 52(Issue 45) pp:
Publication Date(Web):October 22, 2013
DOI:10.1021/bi4012385
EmrD is the only structurally characterized drug/H+ antiporter of the major facilitator superfamily (MFS). It has been crystallized in a doubly occluded conformation that is considered representative of an intermediate state in the transport cycle of MFS transporters. However, unexpected features of the crystal structure and the lack of functional information available for EmrD limit the utility of the structural data. To assess whether the crystal structure represents a stable state in a native-like environment, we used electron paramagnetic resonance (EPR) spectroscopy to determine the mobility and accessibility of spin labels at 76 positions in six transmembrane (TM) helices of EmrD reconstituted in liposomes. While the EPR data were mostly consistent with the crystal structure, they also revealed significant deviations from the predicted orientation and topology of TM helices at several locations. Additionally, we were unable to reproduce EmrD-dependent multidrug resistance phenotypes in vitro and in cell-based assays of drug transport. In spite of structural and functional discrepancies, we mapped a pH-dependent conformational change in which the cytoplasmic side of the N-terminal half opened locally in response to protonation. This conformational switch is consistent with the expected pH-dependent behavior of MFS H+-coupled antiporters.
Co-reporter:Ezelle T. McDonald, Marco Bortolus, Hanane A. Koteiche, and Hassane S. Mchaourab
Biochemistry 2012 Volume 51(Issue 6) pp:
Publication Date(Web):January 20, 2012
DOI:10.1021/bi2017624
Human small heat shock protein 27 (Hsp27) undergoes concentration-dependent equilibrium dissociation from an ensemble of large oligomers to a dimer. This phenomenon plays a critical role in Hsp27 chaperone activity in vitro enabling high affinity binding to destabilized proteins. In vivo dissociation, which is regulated by phosphorylation, controls Hsp27 role in signaling pathways. In this study, we explore the sequence determinants of Hsp27 dissociation and define the structural basis underlying the increased affinity of Hsp27 dimers to client proteins. A systematic cysteine mutagenesis is carried out to identify residues in the N-terminal domain important for the equilibrium between Hsp27 oligomers and dimers. In addition, spin-labels were attached to the cysteine mutants to enable electron paramagnetic resonance (EPR) analysis of residue environment and solvent accessibility in the context of the large oligomers, upon dissociation to the dimer, and following complex formation with the model substrate T4 Lysozyme (T4L). The mutagenic analysis identifies residues that modulate the equilibrium dissociation in favor of the dimer. EPR analysis reveals that oligomer dissociation disrupts subunit contacts leading to the exposure of Hsp27 N-terminal domain to the aqueous solvent. Moreover, regions of this domain are highly dynamic with no evidence of a packed core. Interaction between T4L and sequences in this domain is inferred from transition of spin-labels to a buried environment in the substrate/Hsp27 complex. Together, the data provide the first structural analysis of sHSP dissociation and support a model of chaperone activity wherein unstructured and highly flexible regions in the N-terminal domain are critical for substrate binding.
Co-reporter:Hassane S. Mchaourab, Yi-Lun Lin, and Benjamin W. Spiller
Biochemistry 2012 Volume 51(Issue 25) pp:5105-5112
Publication Date(Web):June 6, 2012
DOI:10.1021/bi300525x
How does the sequence of a single small heat shock protein (sHSP) assemble into oligomers of different sizes? To gain insight into the underlying structural mechanism, we determined the crystal structure of an engineered variant of Methanocaldococcus jannaschii Hsp16.5 wherein a 14 amino acid peptide from human heat shock protein 27 (Hsp27) was inserted at the junction of the N-terminal region and the α-crystallin domain. In response to this insertion, the oligomer shell expands from 24 to 48 subunits while maintaining octahedral symmetry. Oligomer rearrangement does not alter the fold of the conserved α-crystallin domain nor does it disturb the interface holding the dimeric building block together. Rather, the flexible C-terminal tail of Hsp16.5 changes its orientation relative to the α-crystallin domain which enables alternative packing of dimers. This change in orientation preserves a peptide-in-groove interaction of the C-terminal tail with an adjacent β-sandwich, thereby holding the assembly together. The interior of the expanded oligomer, where substrates presumably bind, retains its predominantly nonpolar character relative to the outside surface. New large windows in the outer shell provide increased access to these substrate-binding regions, thus accounting for the higher affinity of this variant to substrates. Oligomer polydispersity regulates sHSPs chaperone activity in vitro and has been implicated in their physiological roles. The structural mechanism of Hsp16.5 oligomer flexibility revealed here, which is likely to be highly conserved across the sHSP superfamily, explains the relationship between oligomer expansion observed in disease-linked mutants and changes in chaperone activity.
Co-reporter:Joey C. Latham, Richard A. Stein, Darryl J. Bornhop and Hassane S. Mchaourab
Analytical Chemistry 2009 Volume 81(Issue 5) pp:1865
Publication Date(Web):January 29, 2009
DOI:10.1021/ac802327h
We report the quantitative, label-free analysis of protein−protein interactions in free solution within picoliter volumes using backscatter interferometry (BSI). Changes in the refractive index are measured for solutions introduced on a PDMS microchip allowing determination of forward and reverse rate constants for two-mode binding. Time-dependent BSI traces are directly fit using a global analysis approach to characterize the interaction of the small heat-shock protein α-Crystallin with two substrates: destabilized mutants of T4 lysozyme and the in vivo target βB1-Crystallin. The results recapitulate the selectivity of αB-Crystallin differentially binding T4L mutants according to their free energies of unfolding. Furthermore, we demonstrate that an αA-Crystallin mutant linked to hereditary cataract has activated binding to βB1-Crystallin. Binding isotherms obtained from steady-state values of the BSI signal yielded meaningful dissociation constants and establishes BSI as a novel tool for the rapid identification of molecular partners using exceedingly small sample quantities under physiological conditions. This work demonstrates that BSI can be extended to screen libraries of disease-related mutants to quantify changes in affinity and/or kinetics of binding.
Co-reporter:Hassane S. Mchaourab, Jared A. Godar and Phoebe L. Stewart
Biochemistry 2009 Volume 48(Issue 18) pp:
Publication Date(Web):March 26, 2009
DOI:10.1021/bi900212j
Small heat shock proteins (sHSP) make up a remarkably diverse group of molecular chaperones possessing a degree of structural plasticity unparalleled in other protein superfamilies. In the absence of chemical energy input, these stability sensors can sensitively recognize and bind destabilized proteins, even in the absence of gross misfolding. Cellular conditions regulate affinity toward client proteins, allowing tightly controlled switching and tuning of sHSP chaperone capacity. Perturbations of this regulation, through chemical modification or mutation, directly lead to a variety of disease states. This review explores the structural basis of sHSP oligomeric flexibility and the corresponding functional consequences in the context of a model describing sHSP activity with a set of three coupled thermodynamic equilibria. As current research illuminates many novel physiological roles for sHSP outside of their traditional duties as molecular chaperones, such a conceptual framework provides a sound foundation for describing these emerging functions in physiological and pathological processes.
Co-reporter:Hassane S. Mchaourab, Sanjay Mishra, Hanane A. Koteiche and Sepan H. Amadi
Biochemistry 2008 Volume 47(Issue 31) pp:
Publication Date(Web):July 11, 2008
DOI:10.1021/bi800628d
EmrE is the prototype of small multidrug resistance transporters and has emerged as a model of membrane protein evolution. Analysis of the distances separating symmetry-related site-specific spin labels, correlation of topological sequence bias to C-terminal orientation, to membrane insertion efficiency, and to resistance to ethidium bromide collectively demonstrate that EmrE monomers adopt a parallel topology in the functional dimer. We propose a coupled insertion and assembly model for EmrE in which the favorable energetics of the parallel dimer interface override topological constraints arising from weak asymmetry in positive charge distribution.
Co-reporter:Jinhui Dong;Guangyong Yang
Science 2005 Vol 308(5724) pp:1023-1028
Publication Date(Web):13 May 2005
DOI:10.1126/science.1106592
Abstract
We used site-directed spin-labeling and electron paramagnetic resonance spectroscopy to characterize the conformational motion that couples energy expenditure to substrate translocation in the multidrug transporter MsbA. In liposomes, ligand-free MsbA samples conformations that depart from the crystal structures, including looser packing and water penetration along the periplasmic side. Adenosine triphosphate (ATP) binding closes the substrate chamber to the cytoplasm while increasing hydration at the periplasmic side, consistent with an alternating access model. Accentuated by ATP hydrolysis, the changes in the chamber dielectric environment and its geometry provide the likely driving force for flipping amphipathic substrates and a potential exit pathway. These results establish the structural dynamic basis of the power stroke in multidrug-resistant ATP-binding cassette (MDR ABC) transporters.
Co-reporter:Ping Zou, Shu-Yu Wu, Hanane A. Koteiche, Sanjay Mishra, Daniel S. Levic, Ela Knapik, Wenbiao Chen, Hassane S. Mchaourab
Experimental Eye Research (September 2015) Volume 138() pp:104-113
Publication Date(Web):1 September 2015
DOI:10.1016/j.exer.2015.07.001
•We generated and characterized the first zebrafish knockout mutant for αA-crystallin.•Loss of αA-crystallin severely affects zebrafish embryonic lens morphology.•Maternal expression of αA-crystallin plays a role in early zebrafish lens development.•The role of αA-crystallin in lens development is conserved in vertebrates.•Our results demonstrate a relevant usage of zebrafish lens to study α-crystallin functions in vivo.αA- and αB-crystallins are small heat shock proteins that bind thermodynamically destabilized proteins thereby inhibiting their aggregation. Highly expressed in the mammalian lens, the α-crystallins have been postulated to play a critical role in the maintenance of lens optical properties by sequestering age-damaged proteins prone to aggregation as well as through a multitude of roles in lens epithelial cells. Here, we have examined the role of α-crystallins in the development of the vertebrate zebrafish lens. For this purpose, we have carried out morpholino-mediated knockdown of αA-, αBa- and αBb-crystallin and characterized the gross morphology of the lens. We observed lens abnormalities, including increased reflectance intensity, as a consequence of the interference with expression of these proteins. These abnormalities were less frequent in transgenic zebrafish embryos expressing rat αA-crystallin suggesting a specific role of α-crystallins in embryonic lens development. To extend and confirm these findings, we generated an αA-crystallin knockout zebrafish line. A more consistent and severe lens phenotype was evident in maternal/zygotic αA-crystallin mutants compared to those observed by morpholino knockdown. The penetrance of the lens phenotype was reduced by transgenic expression of rat αA-crystallin and its severity was attenuated by maternal αA-crystallin expression. These findings demonstrate that the role of α-crystallins in lens development is conserved from mammals to zebrafish and set the stage for using the embryonic lens as a model system to test mechanistic aspects of α-crystallin chaperone activity and to develop strategies to fine-tune protein–protein interactions in aging and cataracts.
Co-reporter:Kelli Kazmier, Nathan S. Alexander, Jens Meiler, Hassane S. Mchaourab
Journal of Structural Biology (March 2011) Volume 173(Issue 3) pp:549-557
Publication Date(Web):1 March 2011
DOI:10.1016/j.jsb.2010.11.003
A hybrid protein structure determination approach combining sparse Electron Paramagnetic Resonance (EPR) distance restraints and Rosetta de novo protein folding has been previously demonstrated to yield high quality models (Alexander et al. (2008)). However, widespread application of this methodology to proteins of unknown structures is hindered by the lack of a general strategy to place spin label pairs in the primary sequence. In this work, we report the development of an algorithm that optimally selects spin labeling positions for the purpose of distance measurements by EPR. For the α-helical subdomain of T4 lysozyme (T4L), simulated restraints that maximize sequence separation between the two spin labels while simultaneously ensuring pairwise connectivity of secondary structure elements yielded vastly improved models by Rosetta folding. 54% of all these models have the correct fold compared to only 21% and 8% correctly folded models when randomly placed restraints or no restraints are used, respectively. Moreover, the improvements in model quality require a limited number of optimized restraints, which is determined by the pairwise connectivities of T4L α-helices. The predicted improvement in Rosetta model quality was verified by experimental determination of distances between spin labels pairs selected by the algorithm. Overall, our results reinforce the rationale for the combined use of sparse EPR distance restraints and de novo folding. By alleviating the experimental bottleneck associated with restraint selection, this algorithm sets the stage for extending computational structure determination to larger, traditionally elusive protein topologies of critical structural and biochemical importance.
Co-reporter:Hassane S. Mchaourab, P. Ryan Steed, Kelli Kazmier
Structure (9 November 2011) Volume 19(Issue 11) pp:1549-1561
Publication Date(Web):9 November 2011
DOI:10.1016/j.str.2011.10.009
Trapping membrane proteins in the confines of a crystal lattice obscures dynamic modes essential for interconversion between multiple conformations in the functional cycle. Moreover, lattice forces could conspire with detergent solubilization to stabilize a minor conformer in an ensemble thus confounding mechanistic interpretation. Spin labeling in conjunction with electron paramagnetic resonance (EPR) spectroscopy offers an exquisite window into membrane protein dynamics in the native-like environment of a lipid bilayer. Systematic application of spin labeling and EPR identifies sequence-specific secondary structures, defines their topology and their packing in the tertiary fold. Long range distance measurements (60 Å–80 Å) between pairs of spin labels enable quantitative analysis of equilibrium dynamics and triggered conformational changes. This review highlights the contribution of spin labeling to bridging structure and mechanism. Efforts to develop methods for determining structures from EPR restraints and to increase sensitivity and throughput promise to expand spin labeling applications in membrane protein structural biology.
Co-reporter:Ping Zou, Hassane S. Mchaourab
Journal of Molecular Biology (30 October 2009) Volume 393(Issue 3) pp:574-585
Publication Date(Web):30 October 2009
DOI:10.1016/j.jmb.2009.08.051
MsbA is an ATP-binding cassette transporter from Escherichia coli that is involved in trafficking lipid A across the inner membrane. ATP-binding cassette transporters harness the free energy of ATP binding and hydrolysis to drive the uphill translocation of substrates against their concentration gradients. A model of protein motion coupling energy input to work was inspired by crystallographic snapshots of MsbA. Central to this model is a switch in the accessibility of a transmembrane chamber, implicated in substrate binding, from an inward- to an outward-facing configuration. Here, we used spin labeling and electron paramagnetic resonance spectroscopy to systematically explore rearrangements in MsbA structure during the ATP hydrolysis cycle. Spin-label accessibility and local dynamics were determined in liposomes for the nucleotide-free intermediate and the transition state of ATP hydrolysis. The changes in the electron paramagnetic resonance parameters between these two intermediates fit a global pattern consistent with alternating access of the chamber. In the transition state of ATP hydrolysis, spin labels on the cytoplasmic side report increased dynamic restrictions and reduced water accessibility, while those on the extracellular side report increased water penetration. Furthermore, spin-label mobility and accessibility as well as their changes are consistent with those expected based on the crystal structures. The reversal in chamber hydration is likely to reduce the free energy of amphipathic substrate binding and promote translocation across the membrane.
Co-reporter:Ping Zou, Marco Bortolus, Hassane S. Mchaourab
Journal of Molecular Biology (30 October 2009) Volume 393(Issue 3) pp:586-597
Publication Date(Web):30 October 2009
DOI:10.1016/j.jmb.2009.08.050
Driven by the energy of ATP binding and hydrolysis, ATP-binding cassette transporters alternate between inward- and outward-facing conformations, allowing vectorial movement of substrates. Conflicting models have been proposed to describe the conformational motion underlying this switch in access of the transport pathway. One model, based on three crystal structures of the lipid flippase MsbA, envisions a large-amplitude motion that disengages the nucleotide-binding domains and repacks the transmembrane helices. To test this model and place the crystal structures in a mechanistic context, we use spin labeling and double electron–electron resonance spectroscopy to define the nature and amplitude of MsbA conformational change during ATP hydrolysis cycle. For this purpose, spin labels were introduced at sites selected to provide a distinctive pattern of distance changes unique to the crystallographic transformation. Distance changes in liposomes, induced by the transition from nucleotide-free MsbA to the highest energy intermediate, fit a simple pattern whereby residues on the cytoplasmic side undergo 20–30 Å closing motion while a 7- to 10-Å opening motion is observed on the extracellular side. The transmembrane helices undergo relative movement to create the outward opening consistent with that implied by the crystal structures. Double electron–electron resonance distance distributions reveal asymmetric backbone flexibility on the two sides of the transporter that correlates with asymmetric opening of the substrate-binding chamber. Together with extensive accessibility analysis, our results suggest that these structures capture features of the motion that couples ATP energy expenditure to work, providing a framework for the mechanism of substrate transport.
Co-reporter:Robel B. Yirdaw, Hassane S. Mchaourab
Biophysical Journal (3 October 2012) Volume 103(Issue 7) pp:
Publication Date(Web):3 October 2012
DOI:10.1016/j.bpj.2012.07.053
Bacteriophage T4 Lysozyme (T4L) catalyzes the hydrolysis of the peptidoglycan layer of the bacterial cell wall late in the infection cycle. It has long been postulated that equilibrium dynamics enable substrate access to the active site located at the interface between the N- and C-terminal domains. Crystal structures of WT-T4L and point mutants captured a range of conformations that differ by the hinge-bending angle between the two domains. Evidence of equilibrium between open and closed conformations in solution was gleaned from distance measurements between the two domains but the nature of the equilibrium and the timescale of the underlying motion have not been investigated. Here, we used fluorescence fluctuation spectroscopy to directly detect T4L equilibrium conformational fluctuations in solution. For this purpose, Tetramethylrhodamine probes were introduced at pairs of cysteines in regions of the molecule that undergo relative displacement upon transition from open to closed conformations. Correlation analysis of Tetramethylrhodamine intensity fluctuations reveals hinge-bending motion that changes the relative distance and orientation of the N- and C-terminal domains with ≅15 μs relaxation time. That this motion involves interconversion between open and closed conformations was further confirmed by the dampening of its amplitude upon covalent substrate trapping. In contrast to the prevalent two-state model of T4L equilibrium, molecular brightness and number of particles obtained from cumulant analysis suggest that T4L populates multiple intermediate states, consistent with the wide range of hinge-bending angles trapped in the crystal structure of T4L mutants.
Co-reporter:Derek P. Claxton, Ping Zou, Hassane S. Mchaourab
Journal of Molecular Biology (25 January 2008) Volume 375(Issue 4) pp:1026-1039
Publication Date(Web):25 January 2008
DOI:10.1016/j.jmb.2007.11.014
We have determined the structural changes that accompany the formation of a stable complex between a destabilized mutant of T4 lysozyme (T4L) and the small heat shock protein α-crystallin. Using pairs of fluorescence or spin label probes to fingerprint the T4L tertiary fold, we demonstrate that binding disrupts tertiary packing in the two domains as well as across the active-site cleft. Furthermore, increased distances between i and i + 4 residues of helices support a model in which the bound structure is not native-like but significantly unfolded. In the confines of the oligomer, T4L has a preferential orientation with residues in the more hydrophobic C-terminal domain sequestered in a buried environment, while residues in the N-terminal domain are exposed to the aqueous solvent. Furthermore, electron paramagnetic resonance spectral line shapes of sites in the N-terminal domain are narrower than in the folded, unbound T4L reflecting an unstructured backbone and an asymmetric pattern of contacts between T4L and α-crystallin. The net orientation is not affected by the location of the destabilizing mutation consistent with the notion that binding is not triggered by recognition of localized unfolding. Together, the structural and thermodynamic data indicate that the stably bound conformation of T4L is unfolded and support a model in which the two modes of substrate binding originate from two discrete binding sites on the chaperone.
Co-reporter:Ping Zou, Hassane S. Mchaourab
Biophysical Journal (17 March 2010) Volume 98(Issue 6) pp:
Publication Date(Web):17 March 2010
DOI:10.1016/j.bpj.2009.12.4193
We report a significant methodological advance in the application of double electron-electron resonance (DEER) spectroscopy to measure long-range distances in spin-labeled membrane proteins. In the pseudo two-dimensional environment of proteoliposomes, a steep intermolecular background shapes DEER signals leading to long accumulation times, complicating data analysis and reducing the maximal measurable distances from 70 Å down to ∼40–50 Å. To eliminate these limitations, we took advantage of the homogeneity and monodispersity of a class of discoidal nanoscale phospholipid bilayers in conjunction with the micromolar DEER sensitivity at Q-band (34 GHz) microwave frequency. Spin-labeled mutants of the ABC transporter MsbA were functionally reconstituted at a ratio of one functional dimer per nanoscale apolipoprotein-bound bilayer (NABB). DEER echo intensities from NABB-reconstituted MsbA have linear baselines reflecting a three-dimensional spatial distribution. This results in an order-of-magnitude higher sensitivity at Q-band relative to proteoliposomes and restores the maximal observable distance effectively increasing experimental throughput. The advances described here set the stage for the use of DEER spectroscopy to analyze conformational dynamics of sample-limited eukaryotic membrane proteins.
Co-reporter:Hassane S. Mchaourab, M. Satish Kumar, Hanane A. Koteiche
FEBS Letters (15 May 2007) Volume 581(Issue 10) pp:1939-1943
Publication Date(Web):15 May 2007
DOI:10.1016/j.febslet.2007.04.005
To elucidate the structural and energetic basis of attractive protein interactions in the aging lens, we investigated the binding of destabilized mutants of βB1-crystallin to the lens chaperones, α-crystallins. We show that the mutations enhance the binding affinity to αA- but not αB-crystallin at physiological temperatures. Complex formation disrupts the dimer interface of βB1-crystallin consistent with the binding of a monomer. Binding isotherms obtained at increasing concentrations of βB1-crystallin deviate from a classic binding equilibrium and display cooperative-like behavior. In the context of βB1-crystallin unfolding equilibrium, these characteristics are reflective of the concentration-dependent change in the population of a dimeric intermediate that has low affinity to αA-crystallin. In the lens, where α-crystallin binding sites are not regenerated, this may represent an added mechanism to maintain lens transparency.
Co-reporter:Hanane A. Koteiche, M. Satish Kumar, Hassane S. Mchaourab
FEBS Letters (15 May 2007) Volume 581(Issue 10) pp:1933-1938
Publication Date(Web):15 May 2007
DOI:10.1016/j.febslet.2007.04.004
A central step in understanding lens aging is to characterize the thermodynamic stability of its proteins and determine the consequences of changes in the primary sequence on their folding equilibria. For this purpose, destabilized mutations were introduced in βB1-crystallin targeting the domain interface within the fold of a subunit. Global unfolding was monitored by tryptophan fluorescence while concomitant structural changes at the dimer interface were monitored by fluorescence and spin labels. Both spectral probes report explicit evidence of multi-state unfolding equilibrium. The biphasic nature of the unfolding curves was more pronounced at higher protein concentration. Distinct shifts in the midpoint of the second transition reflect the population of a dimeric intermediate. This intermediate may be a critical determinant for the life-long stability of the β-crystallins and has important consequences on interactions with α-crystallin.
Co-reporter:Ping Zou, Kavitha Surendhran, Hassane S. Mchaourab
Biophysical Journal (15 February 2007) Volume 92(Issue 4) pp:
Publication Date(Web):15 February 2007
DOI:10.1529/biophysj.106.098913
We demonstrate the feasibility and practical limitations of using steady-state anisotropy to determine distances from fluorescence homotransfer in the context of a protein of known crystal structure. Eight double mutants of T4 lysozyme spanning the distance range between 20 Å and 50 Å were labeled with a methanethiosulfonate derivative of fluorescein. The measured distances in liquid solution are in agreement with those determined from dipolar coupling between spin labels in the frozen state. They can be interpreted in the context of the crystal structure after accounting for the probe linking arm. Overall, the results establish the necessary calibration for this spectroscopic ruler. The measurement of similar distance trends using independent probes sets the stage for the complementary use of homotransfer and dipolar coupling in the determination of static structures and detection of conformational changes.
.jpg)
![1-Piperidinyloxy,4-[2-[1-[(2S,4S)-4-[(3-amino-2,3,6-trideoxy-a-L-lyxo-hexopyranosyl)oxy]-1,2,3,4,6,11-hexahydro-2,5,12-trihydroxy-7-methoxy-6,11-dioxo-2-naphthacenyl]ethylidene]hydrazinylidene]-2,2,6,6-tetramethyl-](http://img.cochemist.com/ccimg/84500/84412-94-2.png)
![1-Piperidinyloxy,4-[2-[1-[(2S,4S)-4-[(3-amino-2,3,6-trideoxy-a-L-lyxo-hexopyranosyl)oxy]-1,2,3,4,6,11-hexahydro-2,5,12-trihydroxy-7-methoxy-6,11-dioxo-2-naphthacenyl]ethylidene]hydrazinylidene]-2,2,6,6-tetramethyl-](http://img.cochemist.com/ccimg/84500/84412-94-2_b.png)
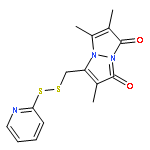
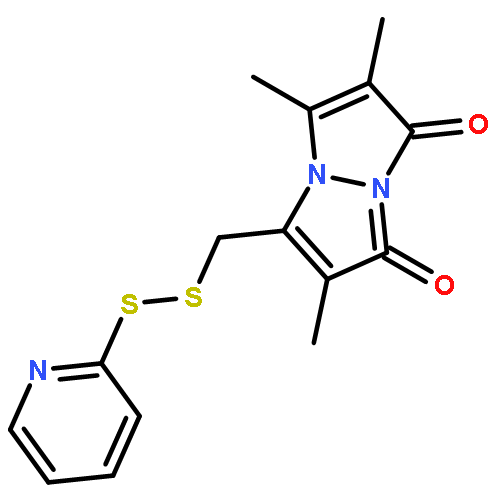


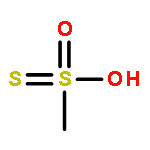
![1H-Pyrrol-1-yloxy,2,5-dihydro-2,2,5,5-tetramethyl-3-[[(methylsulfonyl)thio]methyl]-](http://img.cochemist.com/ccimg/81300/81213-52-7.png)
![1H-Pyrrol-1-yloxy,2,5-dihydro-2,2,5,5-tetramethyl-3-[[(methylsulfonyl)thio]methyl]-](http://img.cochemist.com/ccimg/81300/81213-52-7_b.png)
![1-Pyrrolidinyloxy,3-[(2-iodoacetyl)amino]-2,2,5,5-tetramethyl-](http://img.cochemist.com/ccimg/27100/27048-01-7.png)
![1-Pyrrolidinyloxy,3-[(2-iodoacetyl)amino]-2,2,5,5-tetramethyl-](http://img.cochemist.com/ccimg/27100/27048-01-7_b.png)
![(2-ISOPROPYL-3-INDOLIZINYL)(4-{3-[(2-METHYL-2-PROPANYL)AMINO]PROP<WBR />OXY}PHENYL)METHANONE](http://img.cochemist.com/ccimg/3600/3546-21-2.png)
![(2-ISOPROPYL-3-INDOLIZINYL)(4-{3-[(2-METHYL-2-PROPANYL)AMINO]PROP<WBR />OXY}PHENYL)METHANONE](http://img.cochemist.com/ccimg/3600/3546-21-2_b.png)