Co-reporter:Yitzhak Tor
Chem 2017 Volume 2, Issue 4(Volume 2, Issue 4) pp:
Publication Date(Web):13 April 2017
DOI:10.1016/j.chempr.2017.03.018
Download high-res image (394KB)Download full-size imageYitzhak Tor is a professor of chemistry and biochemistry and the George W. and Carol A. Lattimer Professor at the University of California, San Diego. He earned his doctorate degree at the Weizmann Institute of Science (1990) and was a postdoctoral fellow at the California Institute of Technology (1990–1993). His research focuses on the chemistry and biology of nucleosides, nucleotides, and nucleic acids as well as the development of cellular delivery agents and fluorescent nucleoside analogs. He is the editor-in-chief of Perspectives in Medicinal Chemistry and Organic Chemistry Insights.
Co-reporter:Ryan J. Weiss;Jeffrey D. Esko
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 27) pp:5656-5668
Publication Date(Web):2017/07/12
DOI:10.1039/C7OB01058C
Heparin and heparan sulfate glycosaminoglycans are long, linear polysaccharides that are made up of alternating dissacharide sequences of sulfated uronic acid and amino sugars. Unlike heparin, which is only found in mast cells, heparan sulfate is ubiquitously expressed on the cell surface and in the extracellular matrix of all animal cells. These negatively-charged glycans play essential roles in important cellular functions such as cell growth, adhesion, angiogenesis, and blood coagulation. These biomolecules are also involved in pathophysiological conditions such as pathogen infection and human disease. This review discusses past and current methods for targeting these complex biomolecules as a novel therapeutic strategy to treating disorders such as cancer, neurodegenerative diseases, and infection.
Co-reporter:Patrycja A. Hopkins;Lisa S. McCoy
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 3) pp:684-690
Publication Date(Web):2017/01/18
DOI:10.1039/C6OB02080A
To display favorable fluorescent properties, the non-emissive native nucleosides need to be modified. Here we present a motif that relies on conjugating 5-membered aromatic heterocycles (e.g., thiophene) to a 6-azapyrimidine (1,2,4-triazine) core. Synthetic accessibility and desirable photophysical properties make these nucleosides attractive candidates for enzymatic incorporation and biochemical assays. While 6-azauridine triphosphate is known to be poorly tolerated by polymerases in RNA synthesis, we illustrate that conjugating a thiophene ring at position 5 overcomes such limitations, facilitating its T7 RNA polymerase-mediated in vitro transcription incorporation into RNA constructs. We further show that the modified transcripts can be ligated to longer oligonucleotides to form singly modified RNAs, as illustrated for an A-site hairpin model RNA construct, which was employed to visualize aminoglycoside antibiotics binding.
Co-reporter:Alexander R. Rovira;Andrea Fin
Chemical Science (2010-Present) 2017 vol. 8(Issue 4) pp:2983-2993
Publication Date(Web):2017/03/28
DOI:10.1039/C6SC05354H
A series of emissive ribonucleoside purine mimics, all comprised of an isothiazolo[4,3-d]pyrimidine core, was prepared using a divergent pathway involving a key Thorpe–Ziegler cyclization. In addition to an adenosine and a guanosine mimic, analogues of the noncanonical xanthosine, isoguanosine, and 2-aminoadenosine were also synthesized and found to be emissive. Isothiazolo 2-aminoadenosine, an adenosine surrogate, was found to be particularly emissive and effectively deaminated by adenosine deaminase. Competitive studies with adenosine deaminase with each analogue in combination with native adenosine showed preference for the native substrate while still deaminating the isothiazolo analogues.
Co-reporter:Kristina M. Hamill, Lisa S. McCoy, Ezequiel Wexselblatt, Jeffrey D. Esko and Yitzhak Tor
Chemical Science 2016 vol. 7(Issue 8) pp:5059-5068
Publication Date(Web):25 Apr 2016
DOI:10.1039/C6SC00488A
Polymyxin B is an antibiotic used against multi-resistant Gram negative infections, despite observed nephrotoxicity. Here we report the synthesis of functionalized derivatives of polymyxin B and its per-guanidinylated derivative in order to further explore the structural requirements necessary to facilitate uptake of the antibiotic into mammalian cells. We also investigate the possibility of using these novel scaffolds as molecular transporters. At nanomolar concentrations, both are capable of delivering large cargo (>300 kDa) into living cells. Their uptake depends exclusively on cell surface heparan sulfate. Mechanistic studies indicate these novel transporters are internalized through caveolae-mediated pathways and confocal microscopy show colocalization with lysosomes. The polymyxin-based transporters demonstrate cytosolic delivery through the delivery of a ribosome-inactivating protein. Furthermore, the natural polymyxin scaffold can be incorporated into liposomes and enhance their intracellular uptake. In addition to demonstrating the ability of the polymyxin scaffold to facilitate internalization into mammalian cells, these observations suggest the potential use of polymyxin and guanidinopolymyxin for intracellular delivery.
Co-reporter:C. Vranken, A. Fin, P. Tufar, J. Hofkens, M. D. Burkart and Y. Tor
Organic & Biomolecular Chemistry 2016 vol. 14(Issue 26) pp:6189-6192
Publication Date(Web):24 May 2016
DOI:10.1039/C6OB00844E
SalL, an enzyme that catalyzes the synthesis of SAM from L-methionine and 5′-chloro-5′-deoxyoadenosine, is shown to accept 5′-chloro-5′-deoxythienoadenosine as a substrate and facilitate the synthesis of a synthetic SAM analog with an unnatural nucleobase. This synthetic cofactor is demonstrated to replace SAM in the DNA methylation reaction with M.TaqI.
Co-reporter:Noam S. Freeman, Curtis E. Moore, L. Marcus Wilhelmsson, and Yitzhak Tor
The Journal of Organic Chemistry 2016 Volume 81(Issue 11) pp:4530-4539
Publication Date(Web):April 29, 2016
DOI:10.1021/acs.joc.6b00310
Nucleodyes, visibly colored chromophoric nucleoside analogues, are reported. Design criteria are outlined and the syntheses of cytidine and uridine azo dye analogues derived from 6-aminouracil are described. Structural analysis shows that the nucleodyes are sound structural analogues of their native nucleoside counterparts, and photophysical studies demonstrate that the nucleodyes are sensitive to microenvironmental changes. Quantum chemical calculations are presented as a valuable complementary tool for the design of strongly absorbing nucleodyes, which overlap with the emission of known fluorophores. Förster critical distance (R0) calculations determine that the nucleodyes make good FRET pairs with both 2-aminopurine (2AP) and pyrrolocytosine (PyC). Additionally, unique tautomerization features exhibited by 5-(4-nitrophenylazo)-6-oxocytidine (8) are visualized by an extraordinary crystal structure.
Co-reporter:Marianna Sholokh;Dr. Roberto Improta;Dr. Mattia Mori;Rajhans Sharma;Dr. Cyril Kenfack;Dr. Dongwon Shin;Dr. Karine Voltz;Dr. Rol H. Stote;Dr. Olga A. Zaporozhets;Dr. Maurizio Botta;Dr. Yitzhak Tor;Dr. Yves Mély
Angewandte Chemie International Edition 2016 Volume 55( Issue 28) pp:7974-7978
Publication Date(Web):
DOI:10.1002/anie.201601688
Abstract
Thienoguanosine (thG) is an isomorphic nucleoside analogue acting as a faithful fluorescent substitute of G, with respectable quantum yield in oligonucleotides. Photophysical analysis of thG reveals the existence of two ground-state tautomers with significantly shifted absorption and emission wavelengths, and high quantum yield in buffer. Using (TD)-DFT calculations, the tautomers were identified as the H1 and H3 keto-amino tautomers. When incorporated into the loop of (−)PBS, the (−)DNA copy of the HIV-1 primer binding site, both tautomers are observed and show differential sensitivity to protein binding. The red-shifted H1 tautomer is strongly favored in matched (−)/(+)PBS duplexes, while the relative emission of the H3 tautomer can be used to detect single nucleotide polymorphisms. These tautomers and their distinct environmental sensitivity provide unprecedented information channels for analyzing G residues in oligonucleotides and their complexes.
Co-reporter:Marianna Sholokh;Dr. Roberto Improta;Dr. Mattia Mori;Rajhans Sharma;Dr. Cyril Kenfack;Dr. Dongwon Shin;Dr. Karine Voltz;Dr. Rol H. Stote;Dr. Olga A. Zaporozhets;Dr. Maurizio Botta;Dr. Yitzhak Tor;Dr. Yves Mély
Angewandte Chemie 2016 Volume 128( Issue 28) pp:8106-8110
Publication Date(Web):
DOI:10.1002/ange.201601688
Abstract
Thienoguanosine (thG) is an isomorphic nucleoside analogue acting as a faithful fluorescent substitute of G, with respectable quantum yield in oligonucleotides. Photophysical analysis of thG reveals the existence of two ground-state tautomers with significantly shifted absorption and emission wavelengths, and high quantum yield in buffer. Using (TD)-DFT calculations, the tautomers were identified as the H1 and H3 keto-amino tautomers. When incorporated into the loop of (−)PBS, the (−)DNA copy of the HIV-1 primer binding site, both tautomers are observed and show differential sensitivity to protein binding. The red-shifted H1 tautomer is strongly favored in matched (−)/(+)PBS duplexes, while the relative emission of the H3 tautomer can be used to detect single nucleotide polymorphisms. These tautomers and their distinct environmental sensitivity provide unprecedented information channels for analyzing G residues in oligonucleotides and their complexes.
Co-reporter:Alexander R. Rovira; Andrea Fin
Journal of the American Chemical Society 2015 Volume 137(Issue 46) pp:14602-14605
Publication Date(Web):November 2, 2015
DOI:10.1021/jacs.5b10420
An evolved fluorescent ribonucleoside alphabet comprising isomorphic purine (tzA, tzG) and pyrimidine (tzU, tzC) analogues, all derived from isothiazolo[4,3-d]pyrimidine as a common heterocyclic core, is described. Structural and biochemical analyses illustrate that the nucleosides, particularly the C-nucleosidic purine analogues, are faithful isomorphic and isofunctional surrogates of their natural counterparts and show improved features when compared to an RNA alphabet derived from thieno[3,4-d]-pyrimidine. The restoration of the nitrogen in a position equivalent to the purines’ N7 leads to “isofunctional” behavior, as illustrated by the ability of adenosine deaminase to deaminate tzA as effectively as adenosine, the native substrate.
Co-reporter:Ryan J. Weiss, Philip L. S. M. Gordts, Dzung Le, Ding Xu, Jeffrey D. Esko and Yitzhak Tor
Chemical Science 2015 vol. 6(Issue 10) pp:5984-5993
Publication Date(Web):29 Jul 2015
DOI:10.1039/C5SC01208B
Surfen, bis-2-methyl-4-amino-quinolyl-6-carbamide, was previously reported as a small molecule antagonist of heparan sulfate (HS), a key cell-surface glycosaminoglycan found on all mammalian cells. To generate structure–activity relationships, a series of rationally designed surfen analogs was synthesized, where its dimeric structure, exocyclic amines, and urea linker region were modified to probe the role of each moiety in recognizing HS. An in vitro assay monitoring inhibition of fibroblast growth factor 2 binding to wild-type CHO cells was utilized to quantify interactions with cell surface HS. The dimeric molecular structure of surfen and its aminoquinoline ring systems was essential for its interaction with HS, and certain dimeric analogs displayed higher inhibitory potency than surfen and were also shown to block downstream FGF signaling in mouse embryonic fibroblast cells. These molecules were also able to antagonize other HS–protein interactions including the binding of soluble RAGE to HS. Importantly, selected molecules were shown to neutralize heparin and other heparinoids, including the synthetic pentasaccharide fondaparinux, in a factor Xa chromogenic assay and in vivo in mice. These results suggest that small molecule antagonists of heparan sulfate and heparin can be of therapeutic potential for the treatment of disorders involving glycosaminoglycan–protein interactions.
Co-reporter:Dongwon Shin, Peter Lönn, Steven F. Dowdy and Yitzhak Tor
Chemical Communications 2015 vol. 51(Issue 9) pp:1662-1665
Publication Date(Web):05 Dec 2014
DOI:10.1039/C4CC08809C
Singly and multiply modified synthetic siRNA oligonucleotides, containing isomorphic surrogate nucleobases, show high interference potency in cell culture, suggesting the highly isomorphic RNA alphabet, based on a thieno[3,4-d]-pyrimidine core, is tolerated well by the cellular silencing machinery.
Co-reporter:Andro C. Rios;Hiu T. Yu
Journal of Physical Organic Chemistry 2015 Volume 28( Issue 3) pp:173-180
Publication Date(Web):
DOI:10.1002/poc.3318
Nature's selection of the contemporary nucleobases in RNA and DNA continues to intrigue the origin of life community. While the prebiotic synthesis of the N-glycosyl bond has historically been a central area of investigation, variations in hydrolytic stability among the N-glycosyl bonds may have presented an additional selection pressure that contributed to nucleobase and nucleoside evolution. To experimentally probe this hypothesis, a systematic kinetic analysis of the hydrolytic deglycosylation reactions of modified, alternative, and native nucleosides was undertaken. Rate constants were measured as a function of temperature (at pH 1) to produce Arrhenius and Eyring plots for extrapolation to 37°C and determination of thermodynamic activation parameters. Rate enhancements based on the differences in reaction rates of deoxyriboglycosidic and riboglycosidic bonds were found to vary under the same conditions. Rate constants of deoxynucleosides were also measured across the pH range of 1–3 (at 50°C), which highlighted how simple changes to the heterocycle alone can lead to a significant variation in deglycosylation rates. The contemporary nucleosides exhibited the slowest deglycosylation rates in comparison with the nonnative/alternative nucleosides, which we suggest as experimental support for nature's selection of the fittest N-glycosyl bonds. Copyright © 2014 John Wiley & Sons, Ltd.
Co-reporter:Ezequiel Wexselblatt, Jeffrey D. Esko, and Yitzhak Tor
ACS Nano 2015 Volume 9(Issue 4) pp:3961
Publication Date(Web):April 1, 2015
DOI:10.1021/nn507382n
GNeosomes, lysosomotropic lipid vesicles decorated with guanidinoneomycin, can encapsulate and facilitate the cellular internalization and lysosomal delivery of cargo ranging from small molecules to high molecular weight proteins, in a process that is exclusively dependent on cell surface glycosaminoglycans. Their cellular uptake mechanism and co-localization with lysosomes, as well as the delivery, release, and activity of internalized cargo, are quantified. GNeosomes are proposed as a universal platform for lysosomal delivery with potential as a basic research tool and a therapeutic vehicle.Keywords: cellular delivery; guanidinoglycosides; liposomes; lysosomes; nanoassemblies;
Co-reporter:Dr. Rena A. Mizrahi;Dr. Dongwon Shin;Dr. Renatus W. Sinkeldam;Dr. Kelly J. Phelps;Dr. Andrea Fin; Dean J. Tantillo; Yitzhak Tor; Peter A. Beal
Angewandte Chemie International Edition 2015 Volume 54( Issue 30) pp:8713-8716
Publication Date(Web):
DOI:10.1002/anie.201502070
Abstract
Adenosine to inosine RNA editing catalyzed by ADAR enzymes is common in humans, and altered editing is associated with disease. Experiments using substrate RNAs with adenosine analogues at editing sites are useful for defining features of the ADAR reaction mechanism. The reactivity of ADAR2 was evaluated with RNA containing the emissive adenosine analogue thieno[3,4-d]-6-aminopyrimidine (thA). This nucleoside was incorporated into a mimic of the glutamate receptor B (GluR B) mRNA R/G editing site. We found that thA is recognized by AMV reverse transcriptase as A, and is deaminated rapidly by human ADAR2 to give thI. Importantly, ADAR reaction progress can be monitored by following the deamination-induced change in fluorescence of the thA-modified RNA. The observed high thA reactivity adds to our understanding of the structural features that are necessary for an efficient hADAR2 reaction. Furthermore, the new fluorescent assay is expected to accelerate mechanistic studies of ADARs.
Co-reporter:Dr. Rena A. Mizrahi;Dr. Dongwon Shin;Dr. Renatus W. Sinkeldam;Dr. Kelly J. Phelps;Dr. Andrea Fin; Dean J. Tantillo; Yitzhak Tor; Peter A. Beal
Angewandte Chemie 2015 Volume 127( Issue 30) pp:8837-8840
Publication Date(Web):
DOI:10.1002/ange.201502070
Abstract
Adenosine to inosine RNA editing catalyzed by ADAR enzymes is common in humans, and altered editing is associated with disease. Experiments using substrate RNAs with adenosine analogues at editing sites are useful for defining features of the ADAR reaction mechanism. The reactivity of ADAR2 was evaluated with RNA containing the emissive adenosine analogue thieno[3,4-d]-6-aminopyrimidine (thA). This nucleoside was incorporated into a mimic of the glutamate receptor B (GluR B) mRNA R/G editing site. We found that thA is recognized by AMV reverse transcriptase as A, and is deaminated rapidly by human ADAR2 to give thI. Importantly, ADAR reaction progress can be monitored by following the deamination-induced change in fluorescence of the thA-modified RNA. The observed high thA reactivity adds to our understanding of the structural features that are necessary for an efficient hADAR2 reaction. Furthermore, the new fluorescent assay is expected to accelerate mechanistic studies of ADARs.
Co-reporter:Lisa S. McCoy ; Dongwon Shin
Journal of the American Chemical Society 2014 Volume 136(Issue 43) pp:15176-15184
Publication Date(Web):September 25, 2014
DOI:10.1021/ja5039227
The fastidious behavior of T7 RNA polymerase limits the incorporation of synthetic nucleosides into RNA transcripts, particularly at or near the promoter. The practically exclusive use of GTP for transcription initiation further compounds this challenge, and reactions with GTP analogs, where the heterocyclic nucleus has been altered, have not, to our knowledge, been demonstrated. The enzymatic incorporation of thGTP, a newly synthesized isomorphic fluorescent nucleotide with a thieno[3,4-d]pyrimidine core, is explored. The modified nucleotide can initiate and maintain transcription reactions, leading to the formation of fully modified and highly emissive RNA transcripts with thG replacing all guanosine residues. Short and long modified transcripts are synthesized in comparable yields to their natural counterparts. To assess proper folding and function, transcripts were used to assemble a hammerhead ribozyme with all permutations of natural and modified enzyme and substrate strands. The thG modified substrate was effectively cleaved by the natural RNA enzyme, demonstrating the isomorphic features of the nucleoside and its ability to replace G residues while retaining proper folding. In contrast, the thG modified enzyme showed little cleavage ability, suggesting the modifications likely disrupted the catalytic center, illustrating the significance of the Hoogsteen face in mediating appropriate contacts. Importantly, the ribozyme cleavage reaction of the emissive fluorescent transcripts could be followed in real time by fluorescence spectroscopy. Beyond their utility as fluorescent probes in biophysical and discovery assays, the results reported point to the potential utility of such isomorphic nucleosides in probing specific mechanistic questions in RNA catalysis and RNA structural analysis.
Co-reporter:Patrycja A. Hopkins, Renatus W. Sinkeldam, and Yitzhak Tor
Organic Letters 2014 Volume 16(Issue 20) pp:5290-5293
Publication Date(Web):October 6, 2014
DOI:10.1021/ol502435d
A family of extended 5-modified-6-aza-uridines was obtained via Suzuki coupling reactions with a common brominated precursor. Extending the conjugated-6-aza-uridines with substituted aryl rings increases the push–pull interactions yielding enhanced bathochromic shifts and solvatochromism compared to the parent nucleosides. For example, the methoxy substituted derivative 1d displays λmax abs around 375 nm, with visible emission maxima at 486 nm (Φ = 0.74) and 525 nm (Φ = 0.02) in dioxane and water, respectively.
Co-reporter: Ulrich F. Müller; Yitzhak Tor
Angewandte Chemie International Edition 2014 Volume 53( Issue 21) pp:5245-5247
Publication Date(Web):
DOI:10.1002/anie.201400847
Co-reporter:Dr. Makoto Inoue;Dr. Ezequiel Wexselblatt; Dr. Jeffrey D. Esko; Dr. Yitzhak Tor
ChemBioChem 2014 Volume 15( Issue 5) pp:676-680
Publication Date(Web):
DOI:10.1002/cbic.201300606
Abstract
Guanidinoglycosides, a family of cellular transporters capable of delivering high Mw biopolymers, have previously been shown to display high selectivity for cell-surface heparan sulfate proteoglycans and promote their clustering. Herein, the internalization mechanism of amphiphilic guanidinoglycoside derivatives was investigated by cell-surface FRET analysis. Unexpectedly, although the heparan sulfate selectivity is maintained, the cellular uptake of these derivatives does not appear to involve clustering of the proteoglycans on the cell surface. This suggests a distinct uptake mechanism when compared to the parent guanidinoglycoside-based carriers.
Co-reporter:Dr. Richard S. K. Lane;Rosemary Jones;Dr. Renatus W. Sinkeldam; Dr. Yitzhak Tor;Dr. Steven W. Magennis
ChemPhysChem 2014 Volume 15( Issue 5) pp:867-871
Publication Date(Web):
DOI:10.1002/cphc.201400031
Abstract
Five isomorphic fluorescent uridine mimics have been subjected to two-photon (2P) excitation analysis to investigate their potential applicability as non-perturbing probes for the single-molecule detection of nucleic acids. We find that small structural differences can cause major changes in the 2P excitation probability, with the 2P cross sections varying by over one order of magnitude. Two of the probes, both thiophene-modified uridine analogs, have the highest 2P cross sections (3.8 GM and 7.6 GM) reported for nucleobase analogs, using a conventional Ti:sapphire laser for excitation at 690 nm; they also have the lowest emission quantum yields. In contrast, the analogs with the highest reported quantum yields have the lowest 2P cross sections. The structure-photophysical property relationship presented here is a first step towards the rational design of emissive nucleobase analogs with controlled 2P characteristics. The results demonstrate the potential for major improvements through judicious structural modifications.
Co-reporter:Ezequiel Wexselblatt, Jeffrey D. Esko, and Yitzhak Tor
The Journal of Organic Chemistry 2014 Volume 79(Issue 15) pp:6766-6774
Publication Date(Web):July 14, 2014
DOI:10.1021/jo501101s
Guanidinium-rich scaffolds facilitate cellular translocation and delivery of bioactive cargos through biological barriers. Although impressive uptake has been demonstrated for nonoligomeric and nonpept(o)idic guanidinylated scaffolds in cell cultures and animal models, the fundamental understanding of these processes is lacking. Charge pairing and hydrogen bonding with cell surface counterparts have been proposed, but their exact role remains putative. The impact of the number and spatial relationships of the guanidinium groups on delivery and organelle/organ localization is yet to be established.
Co-reporter:Dr. Richard J. Fair;Dr. Lisa S. McCoy;Dr. Mary E. Hensler;Bernice Aguilar;Dr. Victor Nizet;Dr. Yitzhak Tor
ChemMedChem 2014 Volume 9( Issue 9) pp:2164-2171
Publication Date(Web):
DOI:10.1002/cmdc.201402175
Abstract
Semisynthetic derivatives of the clinically useful aminoglycosides tobramycin and amikacin were prepared by selectively modifying their 6′′ positions with a variety of hydrogen bond donors and acceptors. Their binding to the rRNA A-site was probed using an in vitro FRET-based assay, and their antibacterial activities against several resistant strains (e.g., Pseudomonas aeruginosa, Klebsiella pneumonia, MRSA) were quantified by determining minimum inhibitory concentrations (MICs). The most potent derivatives were evaluated for their eukaryotic cytotoxicity. Most analogues displayed higher affinity for the bacterial A-site than the parent compounds. Although most tobramycin analogues exhibited no improvement in antibacterial activity, several amikacin analogues showed potent and broad-spectrum antibacterial activity against resistant bacteria. Derivatives tested for eukaryotic cytotoxicity exhibited minimal toxicity, similar to the parent compounds.
Co-reporter:Ludmila Frolov, Andrew Dix, Yitzhak Tor, Alexander B. Tesler, Yulia Chaikin, Alexander Vaskevich, and Israel Rubinstein
Analytical Chemistry 2013 Volume 85(Issue 4) pp:2200
Publication Date(Web):January 31, 2013
DOI:10.1021/ac3029079
RNA is involved in fundamental biological functions when bacterial pathogens replicate. Identifying and studying small molecules that can interact with bacterial RNA and interrupt cellular activities is a promising path for drug design. Aminoglycoside (AMG) antibiotics, prominent natural products that recognize RNA specifically, exert their biological functions by binding to prokaryotic ribosomal RNA and interfering with protein translation, ultimately resulting in bacterial cell death. The decoding site, a small internal loop within the 16S rRNA, is the molecular target for the AMG antibiotics. The specificity of neomycin B, a highly potent AMG antibiotic, to the ribosomal decoding RNA site, was previously studied by observing AMG–RNA complexes in solution. Here, we study this interaction using localized surface plasmon resonance (LSPR) transducers comprising gold island films prepared by evaporation on glass and annealing. Small molecule AMG receptors were immobilized on the Au islands via polyethylene glycol (PEG)-thiol linkers, and the interaction with target RNA in solution was studied by monitoring the change in the LSPR optical response upon binding. The results show high-affinity binding of neomycin to 27-nucleotide model A-site RNA sequence in the nanomolar range, while no specific binding is observed for synthetic RNA oligomers (e.g., poly-U). The impact of specific base substitutions in the A-site RNA constructs on binding affinity and selectivity is determined quantitatively. It is concluded that LSPR is a powerful tool for providing molecular insight into small molecule–RNA interactions and for the design and screening of selective antimicrobial drugs.
Co-reporter:Makoto Inoue, Wenyong Tong, Jeffrey D. Esko, and Yitzhak Tor
ACS Chemical Biology 2013 Volume 8(Issue 7) pp:1383
Publication Date(Web):April 26, 2013
DOI:10.1021/cb400172h
Endocytosis is a key process in cellular delivery of macromolecules by molecular transporters, although the mechanism of internalization remains unclear. Here, we probe the cellular uptake of streptavidin using biotinylated guanidinoneomycin (biotinGNeo), a low molecular weight guanidinium-rich molecular transporter. Two distinct modes were explored: (i) incubation of cells with a preformed tetravalent streptavidin-(biotinGNeo)4 conjugate and (ii) preincubation of cells with the biotinGNeo before exposure to streptavidin. A significant enhancement in uptake was observed after preincubation with biotinGNeo. FRET studies showed that the enhanced uptake was accompanied by extensive aggregation of streptavidin on the cell surface. Because guanidinylated neomycin was previously found to exclusively bind to heparan sulfate, our observations suggest that heparan sulfate proteoglycan aggregation is a pivotal step for endocytic entry into cells by guanidinoglycosides. These observations put forward a practical and general pathway for the cellular delivery of diverse macromolecules.
Co-reporter:Andro C. Rios
Israel Journal of Chemistry 2013 Volume 53( Issue 6-7) pp:469-483
Publication Date(Web):
DOI:10.1002/ijch.201300009
Abstract
The native bases of RNA and DNA are prominent examples of the narrow selection of organic molecules upon which life is based. How did nature “decide” upon these specific heterocycles? Evidence suggests that many types of heterocycles could have been present on the early Earth. It is therefore likely that the contemporary composition of nucleobases is a result of multiple selection pressures that operated during early chemical and biological evolution. The persistence of the fittest heterocycles in the prebiotic environment towards, for example, hydrolytic and photochemical assaults, may have given some nucleobases a selective advantage for incorporation into the first informational polymers. The prebiotic formation of polymeric nucleic acids employing the native bases remains, however, a challenging problem to reconcile. Hypotheses have proposed that the emerging RNA world may have included many types of nucleobases. This is supported by the extensive utilization of non-canonical nucleobases in extant RNA and the resemblance of many of the modified bases to heterocycles generated in simulated prebiotic chemistry experiments. Selection pressures in the RNA world could have therefore narrowed the composition of the nucleic acid bases. Two such selection pressures may have been related to genetic fidelity and duplex stability. Considering these possible selection criteria, the native bases along with other related heterocycles seem to exhibit a certain level of fitness. We end by discussing the strength of the N-glycosidic bond as a potential fitness parameter in the early DNA world, which may have played a part in the refinement of the alphabetic bases.
Co-reporter:Dr. Renatus W. Sinkeldam;Lisa S. McCoy;Dr. Dongwon Shin ;Dr. Yitzhak Tor
Angewandte Chemie International Edition 2013 Volume 52( Issue 52) pp:14026-14030
Publication Date(Web):
DOI:10.1002/anie.201307064
Co-reporter:Dr. Renatus W. Sinkeldam;Lisa S. McCoy;Dr. Dongwon Shin ;Dr. Yitzhak Tor
Angewandte Chemie 2013 Volume 125( Issue 52) pp:14276-14280
Publication Date(Web):
DOI:10.1002/ange.201307064
Co-reporter:Mary S. Noé, Renatus W. Sinkeldam, and Yitzhak Tor
The Journal of Organic Chemistry 2013 Volume 78(Issue 16) pp:8123-8128
Publication Date(Web):July 16, 2013
DOI:10.1021/jo4008964
5-(Thien-2-yl)-2′-deoxyuridine, an isomorphic fluorescent nucleoside analogue, was incorporated into multiple positions within single stranded oligodeoxynucleotides. With minimal impact on duplex stability and overall structure, oligonucleotides containing three identical isomorphic fluorescent nucleosides in alternating or neighboring positions display enhanced, sequence-dependent on-signals for either duplex formation or dissociation.
Co-reporter:Lisa S. McCoy;Dr. Kade D. Roberts; Dr. Roger L. Nation; Dr. Philip E. Thompson;Dr. Tony Velkov; Dr. Jian Li; Dr. Yitzhak Tor
ChemBioChem 2013 Volume 14( Issue 16) pp:2083-2086
Publication Date(Web):
DOI:10.1002/cbic.201300496
Co-reporter:Mary S. Noé, Andro C. Ríos, and Yitzhak Tor
Organic Letters 2012 Volume 14(Issue 12) pp:3150-3153
Publication Date(Web):2017-2-22
DOI:10.1021/ol3012327
The syntheses of four fluorescent nucleoside analogs, related to pyrrolo-C (PyC) and pyrrolo-dC (PydC) through the conjugation or fusion of a thiophene moiety, are described. A thorough photophysical analysis of the nucleosides, in comparison to PyC, is reported.
Co-reporter:Richard J. Fair;Dr. Mary E. Hensler;Wdee Thienphrapa;Quang N. Dam;Dr. Victor Nizet;Dr. Yitzhak Tor
ChemMedChem 2012 Volume 7( Issue 7) pp:1237-1244
Publication Date(Web):
DOI:10.1002/cmdc.201200150
Abstract
The emergence of virulent, drug-resistant bacterial strains coupled with a minimal output of new pharmaceutical agents to combat them makes this a critical time for antibacterial research. Aminoglycosides are a well-studied, highly potent class of naturally occurring antibiotics with scaffolds amenable to modification, and therefore, they provide an excellent starting point for the development of semisynthetic, next-generation compounds. To explore the potential of this approach, we synthesized a small library of aminoglycoside derivatives selectively and minimally modified at one or two positions with a guanidine group replacing the corresponding amine or hydroxy functionality. Most guanidino-aminoglycosides showed increased affinity for the ribosomal decoding rRNA site, the cognate biological target of the natural products, when compared with their parent antibiotics, as measured by an in vitro fluorescence resonance energy transfer (FRET) A-site binding assay. Additionally, certain analogues showed improved minimum inhibitory concentration (MIC) values against resistant bacterial strains, including methicillin-resistant Staphylococcus aureus (MRSA). An amikacin derivative holds particular promise with activity greater than or equal to the parent antibiotic in the majority of bacterial strains tested.
Co-reporter:Dr. Renatus W. Sinkeldam;Patrycja A. Hopkins ; Dr. Yitzhak Tor
ChemPhysChem 2012 Volume 13( Issue 14) pp:3350-3356
Publication Date(Web):
DOI:10.1002/cphc.201200375
Abstract
Optimized facile syntheses and highly desirable spectroscopic properties of two isomorphic fluorescent pyrimidines, comprising a 1,2,4-triazine motif conjugated to a thiophene (1 a) or a furan (1 b), are described. Although structurally related to their 5-modified uridine counterparts, these modified 6-aza-uridines reveal dramatically improved fluorescence properties and a remarkable sensitivity to polarity and pH changes. The thiophene derivative 1 a has an absorption maximum around 335 nm, which upon excitation yields visible emission with a polarity-sensitive maximum and fluorescence quantum yield ranging from 415 nm (Φ=0.8) to 455 nm (Φ=0.2) in dioxane and water, respectively. Nucleoside 1 a also displays susceptibility to acidity. Correlating emission intensity and solution pH yields a pKa value of 6.7–6.9, reasonably close to physiological pH values. The results illustrate that highly sought-after fluorescence features (brightness and responsiveness) are not necessarily the trait of large fluorophores alone, but can be observed with probes that meet stringent isomorphic design criteria.
Co-reporter:Dongwon Shin
Journal of the American Chemical Society 2011 Volume 133(Issue 18) pp:6926-6929
Publication Date(Web):April 15, 2011
DOI:10.1021/ja201397e
A new functional bifacial nucleoside derived from 7-aminopyrimido[4,5-d]pyrimidine-2,4(1H,3H)-dione, a Janus-type nucleobase, has been synthesized and incorporated into DNA oligonucleotides. The nucleobase, having self-complementary H-bonding faces, mimics both T and A and engages in the corresponding Watson–Crick-like base pairs, forming stable duplexes.
Co-reporter:Dongwon Shin ; Renatus W. Sinkeldam
Journal of the American Chemical Society 2011 Volume 133(Issue 38) pp:14912-14915
Publication Date(Web):August 25, 2011
DOI:10.1021/ja206095a
A fluorescent ribonucleoside alphabet consisting of highly emissive purine (thA, thG) and pyrimidine (thU, thC) analogues, all derived from thieno[3,4-d]pyrimidine as the heterocyclic nucleus, is described. Structural and biophysical analyses demonstrated that the emissive analogues are faithful isomorphic nucleoside surrogates. Photophysical analysis established that the nucleosides offer highly desirable qualities, including visible emission, high quantum yield, and responsiveness to environmental perturbations, traits entirely lacking in their native counterparts.
Co-reporter:Dr. Renatus W. Sinkeldam;Paul Marcus;Dmitriy Uchenik ; Dr. Yitzhak Tor
ChemPhysChem 2011 Volume 12( Issue 12) pp:
Publication Date(Web):
DOI:10.1002/cphc.201190060
Co-reporter:Dr. Renatus W. Sinkeldam;Andrea J. Wheat;He Boyaci ; Dr. Yitzhak Tor
ChemPhysChem 2011 Volume 12( Issue 3) pp:567-570
Publication Date(Web):
DOI:10.1002/cphc.201001002
Co-reporter:Dr. Renatus W. Sinkeldam;Paul Marcus;Dmitriy Uchenik ; Dr. Yitzhak Tor
ChemPhysChem 2011 Volume 12( Issue 12) pp:2260-2265
Publication Date(Web):
DOI:10.1002/cphc.201100315
Abstract
Fluorescent nucleoside analogs, commonly used to explore nucleic acid dynamics, recognition and damage, frequently respond to a single environmental parameter. Herein we address the development of chromophores that can simultaneously probe more than one environmental factor while having each associated with a unique spectroscopic signature. We demonstrate that an isomorphic emissive pyridine-modified 2-deoxy-uridine 1, containing multiple sensory elements, responds to changes in acidity, viscosity, and polarity. Protonation of the pyridine moiety (pKa 4.4) leads to enhanced emission (λem=388 nm) and red-shifted absorption spectra (λabs=319 nm), suggesting the formation of an intramolecular hydrogen bond with the neighboring pyrimidine carbonyl. This “locked” conformation can also be mimicked by increasing solvent viscosity, resulting in a stark enhancement of emission quantum yield. Finally, increasing solvent polarity substantially impacts the chromophore’s Stokes shift [from 5.8×103 cm−1 at ET(30)=36.4 kcal mol−1 to 9.3 ×103 cm−1 at ET(30)=63.1 kcal mol−1]. The opposite effect is seen for the impact of solvent polarity of the protonated form. The characteristic photophysical signature induced by each parameter facilitates the exploration of these environmental factors both individually and simultaneously.
Co-reporter:Renatus W. Sinkeldam, Nicholas J. Greco and Yitzhak Tor
Chemical Reviews 2010 Volume 110(Issue 5) pp:2579
Publication Date(Web):March 5, 2010
DOI:10.1021/cr900301e
Co-reporter:Yun Xie ; Tucker Maxson
Journal of the American Chemical Society 2010 Volume 132(Issue 34) pp:11896-11897
Publication Date(Web):August 9, 2010
DOI:10.1021/ja105244t
A new fluorescent ribonucleoside analogue, containing 5-aminoquinazoline-2,4(1H,3H)-dione, acts as a Förster resonance energy transfer acceptor for tryptophan (R0 = 22 Å) and displays visible emission (440 nm). As tryptophan is frequently found at or near the recognition domains of RNA binding proteins, this FRET pair facilitates the study of RNA binding to native proteins and peptides, which is demonstrated here for the HIV-1 Rev association with the Rev Response Element (RRE).
Co-reporter:Yun Xie, Andrew V. Dix and Yitzhak Tor
Chemical Communications 2010 vol. 46(Issue 30) pp:5542-5544
Publication Date(Web):13 May 2010
DOI:10.1039/C0CC00423E
A FRET assembly reports antibiotic affinities to two different RNA targets. A binder was labeled with a fluorophore that acts both as an acceptor for the emissive nucleoside on the bacterial A-site and a donor fluorophore for the terminally-labeled human A-site. Unlabeled drugs were used to dissociate the labeled antibiotic.
Co-reporter:Yun Xie, Tucker Maxson and Yitzhak Tor
Organic & Biomolecular Chemistry 2010 vol. 8(Issue 22) pp:5053-5055
Publication Date(Web):23 Sep 2010
DOI:10.1039/C0OB00413H
A fluorescent nucleobase analogue, 7-aminoquinazoline-2,4-(1H,3H)-dione, is incorporated into a DNA oligonucleotide and senses mismatched pairing by displaying G-specific fluorescence enhancement.
Co-reporter:Andrew V. Dix;Dr. Lucile Fischer;Dr. Stéphane Sarrazin;Christopher P. H. Redgate; Dr. Jeffrey D. Esko; Dr. Yitzhak Tor
ChemBioChem 2010 Volume 11( Issue 16) pp:2302-2310
Publication Date(Web):
DOI:10.1002/cbic.201000399
Abstract
Oligoarginine and guanidinium-rich molecular transporters have been shown to facilitate the intracellular delivery of a diverse range of biologically relevant cargos. Several such transporters have been suggested to interact with cell-surface heparan sulfate proteoglycans as part of their cell-entry pathway. Unlike for other guanidinium-rich transporters, the cellular uptake of guanidinoglycosides at nanomolar concentrations is exclusively heparan sulfate dependent. As distinct cells differ in their expression levels and/or the composition of cell-surface heparan sulfate proteoglycans, one might be able to exploit such differences to selectively target certain cell types. To systematically investigate the nature of their cell-surface interactions, monomeric and dimeric guanidinoglycosides were synthesized by using neomycin, paromomycin, and tobramycin as scaffolds. These transporters differ in the number and 3D arrangement of their guanidinium groups. Their cellular uptake was measured by flow cytometry in wild-type and mutant Chinese hamster ovary cells after the corresponding fluorescent streptavidin–phycoerythrin-Cy5 conjugates had been generated. All derivatives showed negligible uptake in mutant cells lacking heparan sulfate. Decreasing the number of guanidinium groups diminished uptake, but the three dimensional arrangement of these groups was less important for cellular delivery. Whereas conjugates prepared with the monomeric carriers showed significantly reduced uptake in mutant cells expressing heparan sulfate chains with altered patterns of sulfation, conjugates prepared with the dimeric guanidinoglycosides could overcome this deficiency and maintain high levels of uptake in such deficient cells. This finding suggests that cellular uptake depends on the valency of the transporter and both the content and arrangement of the sulfate groups on the cell-surface receptors. Competition studies with chemically desulfated or carboxy-reduced heparin derivatives corroborated these observations. Taken together, these findings show that increasing the valency of the transporters retains heparan sulfate specificity and provides reagents that could distinguish different cell types based on the specific composition of their cell-surface heparan sulfate proteoglycans.
Co-reporter:Matthew J. Belousoff, Bim Graham, Leone Spiccia and Yitzhak Tor
Organic & Biomolecular Chemistry 2009 vol. 7(Issue 1) pp:30-33
Publication Date(Web):30 Oct 2008
DOI:10.1039/B813252F
A number of aminoglycoside antibiotics, and in particular neomycin B, are demonstrated to promote strand cleavage of RNA oligonucleotides (minimised HIV-1 TAR element and prokaryotic ribosomal A-site), by binding and causing sufficient distortion to the RNA backbone to render it more susceptible to intramolecular transesterification.
Co-reporter:SeergazhiG. Srivatsan Dr. Dr.
Chemistry – An Asian Journal 2009 Volume 4( Issue 3) pp:419-427
Publication Date(Web):
DOI:10.1002/asia.200800370
Co-reporter:Seergazhi G. Srivatsan, Haim Weizman and Yitzhak Tor
Organic & Biomolecular Chemistry 2008 vol. 6(Issue 8) pp:1334-1338
Publication Date(Web):10 Mar 2008
DOI:10.1039/B801054D
A highly emissive nucleobase analog, based on a thieno[3,4-d]pyrimidine core, is enzymatically incorporated into RNA oilgonucleotides that function as base discriminating fluorescent probes.
Co-reporter:Renatus W. Sinkeldam Dr.;Nicholas J. Greco Dr. Dr.
ChemBioChem 2008 Volume 9( Issue 5) pp:706-709
Publication Date(Web):
DOI:10.1002/cbic.200700714
Co-reporter:SeergazhiG. Srivatsan Dr.;NicholasJ. Greco Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 35) pp:6661-6665
Publication Date(Web):
DOI:10.1002/anie.200802199
Co-reporter:SeergazhiG. Srivatsan Dr.;NicholasJ. Greco Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 35) pp:
Publication Date(Web):
DOI:10.1002/anie.200890170
Co-reporter:SeergazhiG. Srivatsan Dr.;NicholasJ. Greco Dr.
Angewandte Chemie 2008 Volume 120( Issue 35) pp:6763-6767
Publication Date(Web):
DOI:10.1002/ange.200802199
Co-reporter:SeergazhiG. Srivatsan Dr.;NicholasJ. Greco Dr.
Angewandte Chemie 2008 Volume 120( Issue 35) pp:
Publication Date(Web):
DOI:10.1002/ange.200890224
Co-reporter:
Nature Protocols 2007 2(2) pp:
Publication Date(Web):2007-03-01
DOI:10.1038/nprot.2006.464
When carefully designed and implemented, fluorescent nucleosides and nucleotides can serve as powerful tools for basic research and drug discovery applications that are centered around nucleic acids. Progress in this field has been driven by several fundamental reasons: (i) the natural purines and pyrimidines found in DNA and RNA are practically nonemissive1, 2, 3, (ii) naturally occurring fluorescent bases (e.g., Y37 in tRNAPhe) are typically so severely modified that they cannot generally serve as natural base mimics, and (iii) end labeling with common fluorescent tags (e.g., fluorescein), while providing generic tagging, is not necessarily sensitive to remote binding events. Fluorescent nucleobase analogs that are capable of mimicking the structure and function of the natural bases, but possess emission properties, have therefore emerged in recent years as powerful probes for investigating nucleic acids sequence, structure, dynamics and recognition4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28.Several important features characterize useful emissive nucleobase analogs. These include: (i) high structural similarity to the natural nucleobases, (ii) emission at long wavelengths, preferably in the visible range (this is typically associated with absorption maxima that are red-shifted when compared to the native heterocycles), (iii) sufficient emission quantum efficiency to facilitate their use in real-time fluorescence-based assays and, notably, (iv) susceptibility to the microenvironment, resulting in markedly different photophysical parameters of the chromophore, particularly in emission color or intensity. It is the latter property that is most challenging, yet critical, for fabricating new and useful fluorescent nucleobase probes.How can environmentally responsive nucleosides be used? One of the contemporary directions represents the design of fluorescent deoxynucleosides that can report the identity of the base opposite to them in a double-stranded helix by yielding different emission wavelengths or intensities. Such base-discriminating fluorescent (BDF) nucleosides, when incorporated into oligonucleotide probes, can find uses in DNA sequence identification and single nucleotide polymorphism (SNP) analysis19. A more established application of fluorescent bases, is to use nucleobase analogs to interrogate the position and environment of individual bases within oligonucleotide targets. A classical example is 2-aminopurine (2AP), an adenine analog that forms base pairs with thymine or uracil that are isostructural to the corresponding Watson-Crick base pairs. This adenosine mimic displays minimal emission when stacked with other bases, but, upon unstacking and exposure to aqueous environments, a significantly enhanced emission is observed29, 30, 31, 32, 33. Many biophysical assays have taken advantage of this feature because base flipping is a rather common event in biologically relevant nucleic acids. Selected examples include: (i) 'real-time' monitoring of the hammerhead ribozyme folding, cleavage and inhibition34, 35, (ii) evaluating the dimerization and isomerization of the HIV-1 dimerization initiation site (DIS)36, (iii) examining RNA-protein interactions such as the HIV RRE–Rev binding37, 38, and (iv) studying the binding of aminoglycoside antibiotics to the bacterial decoding site, known as the A-site39, 40.Besides base flipping, nucleic acids also undergo a variety of chemical transformations, including depurination and depyrimidination. These abasic sites can be generated either spontaneously or via enzymatic base excision of damaged nucleosides. Several methods have been developed for detecting the presence of these cytotoxic DNA lesions. Most techniques require irreversible modifications of isolated DNA41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, whereas others use non-natural nucleobase analogs77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89 and only a few use 2AP to investigate the stacking ability of a base opposite an abasic site90, 91, 92. Fluorescent nucleobase analogs that positively sense the presence of abasic sites could provide useful nondestructive tools for the detection of such important DNA defects.We have recently reported that conjugating 5-membered aromatic heterocycles to a pyrimidine core generates a family of responsive fluorescent nucleoside analogs with emission in the visible range93. Specifically, conjugating a furan moiety to the 5-position of 2′-deoxyuridine yielded a nucleoside analog 2 (Fig. 1) that emits above 425 nm, and is sensitive to changes in solvent environment, being fairly emissive in polar and rather nonemissive in apolar solvents. When strategically incorporated into DNA oligonucleotides (see Fig. 2) and hybridized to complementary sequences, this furan-modified pyrimidine analog positively signals the presence of DNA abasic sites, manifested via a sevenfold enhancement in emission of a lesion-containing DNA when compared to the emission of a perfect duplex93.The protocol outlined below provides a step-by-step synthesis of this useful furan-containing nucleoside and its phosphoramidite, the necessary building block for standard solid-phase DNA synthesis94, 95. Detailed procedures for the synthesis (Fig. 3) and purification of a modified oligonucleotide are given. Additionally, this protocol provides researchers with experimental procedures for basic photophysical examination of the fluorescently tagged oligonucleotide.Synthesis of nucleoside 2: Steps 1–7, 25 min; Steps 8–10, 10 min; Step 11, 2 h; Step 12, 5 min; Step 13, 30 min; Step 14, 20 min; Step 15, 20 min (evaporation depends upon the volume and equipment used); Steps 16–18, 30 min; Steps 19–23, 40 min (for one precipitation; but several will be required).Synthesis of nucleoside 3: Steps 24–28, 25 min; Steps 29 and 30, 8 min; Step 31, 8 h; Step 32, 5 min; Step 33, 20 min (evaporation depends upon the volume and equipment used); Steps 34–36, 20 min; Step 37, 30 min; Step 38, 5 min; Step 39, 20 min (evaporation depends upon the volume and equipment used); Steps 40 and 41, 1 min.Synthesis of nucleoside 4: Steps 42–44, 1 h; Steps 45–48, 22 min; Steps 49–51, 15 min; Step 52, 4 h; Step 53, 5 min; Steps 54 and 55, 25 min; Steps 56 and 57, 15 min; Step 58, 20 min (evaporation depends upon the volume and equipment used), Steps 59–61, 20 min; Step 62, 30 min; Step 63, 5 min; Step 64, 20 min (evaporation depends upon the volume and equipment used), Step 65, 1 min.Site-specific incorporation of nucleoside 4: Steps 66–69, 40 min; Step 70, time varies depending upon the DNA synthesizer used; Steps 71–73, 10 min; Steps 74–77, 7 min; Steps 78 and 79, time varies depending upon the DNA synthesizer used; Steps 80–82, 7 min; Steps 83 and 84, 2 h; Steps 85–87, 20 min; Step 88, 24 h; Step 89, 30 min; Step 90, time varies depending upon the technique and instrument used.Purification of DNA oligonucleotide with nucleoside 1 incorporated: Steps 91 and 92, 5 h; Step 93, 30 min; Step 94, 10 min; Steps 95 and 96, 10 min; Steps 97 and 98, 36 h; Steps 99–101, 20 min; Steps 102 and 103, 15 min; Step 104, 15 min; Step 105, time varies depending upon the technique and instrument used.Troubleshooting advice can be found in Table 1.Steady state fluorescence spectra of 5′-GCGATG2GTAGCG-3′ (Fig. 5) were measured in 10 mM sodium phosphate, 100 mM sodium chloride (pH 7.0) on a Horiba Fluoromax-3 luminescence spectrometer (Fig. 5) and a Perkin Elmer LS 50B luminescence spectrometer (Fig. 3). The minimum concentration of a singly modified oligonucleotide containing nucleoside 2 required for adequate fluorescence readings depends upon the sensitivity of the luminescence spectrometer. For example, adequate fluorescence readings using the Horiba Fluoromax-3 are obtained at 0.1 μM (Fig. 5), whereas the lower limit for the Perkin Elmer LS 50B is 0.5 μM.
1H NMR (400 MHz, DMSO-d
6): δ 11.63 (s, 1H), 8.33 (s, 1H), 7.62 (s, 1H), 6.86 (d, J = 2.8 Hz, 1H), 6.52 (m, 1H), 6.22 (t, J = 6.6 Hz, 1H), 5.27 (d, J = 4.4 Hz, 1H), 5.08 (t, J = 4.4 Hz, 1H), 4.28 (m, 1H), 3.84 (m, 1H), 3.61 (m, 2H), 2.18 (t, J = 5.4 Hz, 2H); 13C NMR (100 MHz, DMSO-d
6): δ 160.1, 149.4, 146.5, 141.5, 134.7, 111.6, 107.9, 105.6, 87.6, 84.7, 70.4, 61.1, 40.1; HR-FAB calculated for C13H15N2O6 [M+H]+ 295.0925, found: 295.0928; UV (buffer): λmax = 252 nm (ε = 13,800 cm−1 M−1); λmax = 316 nm (ε = 11,000 cm−1 M−1); ε260 = 13,000 cm−1 M−1.
5′-dimethoxytrityl-5-(fur-2-yl)-2′-deoxythymidine (3).
1H NMR (400 MHz, DMSO-d
6): δ 11.70 (s, 1H), 7.96 (s, 1H), 7.27 – 7.16 (m, 10H), 6.84 – 6.81 (m, 5H), 6.46 – 6.44 (m, 1H), 6.18 (t, J = 6.6 Hz, 1H), 5.36 (d, J = 4.4 Hz, 1H), 4.23 – 4.19 (m, 1H), 3.96 – 3.93 (m, 1H), 3.69 (s, 6H), 3.24 – 3.15 (m, 2H), 2.27–2.24 (m, 2H); 13C NMR (100 MHz, DMSO-d
6): δ 160.1, 158.0, 149.3, 146.1, 144.7, 141.2, 135.5, 133.9, 129.6, 127.7, 127.6, 126.6, 113.1, 111.4, 107.9, 105.6, 85.8, 85.1, 70.3, 63.6, 55.0, 40.2; ESI-MS calculated for C34H32N2NaO8 [M+Na]+ 619.21, found 619.08.
3′-2-cyanoethyldiisopropylphosphoramidite-5′-dimethoxytrityl-5-(fur-2-yl)-2′-deoxythymidine (4)
1H NMR (400 MHz, DMSO-d
6): δ 11.60 (s, 1H), 7.99 (s, 1H), 7.39 – 7.17 (m, 10H), 6.82 – 6.79 (m, 5H), 6.45 – 6.43 (m, 1H), 6.17 (t, J = 6.4 Hz, 1H), 4.50 – 4.44 (m, 1H), 4.11 – 4.08 (m, 1H), 3.69 – 3.39 (m, 10H), 3.24 – 3.19 (m, 2H), 2.64 – 2.61 (m, 2H), 2.46 – 2.34 (m, 2H), 1.23 – 1.08 (m, 12H); 13C NMR (100 MHz, DMSO-d
6): δ 160.1, 158.1, 149.2, 146.1, 144.6, 141.2, 135.3, 134.0, 129.7, 127.7, 127.6, 126.6, 118.7, 113.1, 111.5, 108.0, 105.7, 85.9, 85.2, 72.3, 63.5, 58.5, 55.0, 42.6, 42.5, 40.2, 24.3, 24.3, 24.2, 19.8; 31P NMR (162 MHz, DMSO-d
6, referenced to H3PO4): δ 148.7, 148.3; ESI-MS calculated for C43H49N4NaO9P [M+Na]+ 819.31, found 819.14.
Co-reporter:
Nature Protocols 2007 2(6) pp:
Publication Date(Web):2007-06-14
DOI:10.1038/nprot.2007.222
Responsive fluorescent nucleoside analogues have been extensively applied in basic research and discovery assays1, 2, 3, 4, 5, 6, 7. These fluorescent nucleoside sensors, when strategically placed, can be employed in investigating nucleic acid structure, dynamics and recognition, as these biological events often lead to conformation perturbations that are translated into changes in the microenvironment of the reporter8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20. Useful responsive nucleobases should possess the following desirable features: (i) be isosteric to the natural bases, while maintaining the hydrogen bonding face; (ii) be emissive at long wavelengths, preferably in the visible region; (iii) display sufficient quantum yield when incorporated into nucleic acids for real-time analysis; and (iv) importantly, be sensitive to changes in their microenvironment.A well-characterized and commonly applied nucleoside analogue is 2-aminopurine (2AP), a fluorescent adenine analogue. The emission of 2AP is strongly quenched when stacked with other bases, but significantly enhanced upon unstacking and exposure to more polar environments21, 22, 23, 24. This useful reporting property has been elegantly exploited in designing several biophysical assays for studying RNA folding and recognition. Examples include monitoring the hammerhead ribozyme folding and activity25, 26, examining the dimerization and isomerization of the HIV-1 dimerization initiation site27, evaluating RNA–protein interactions such as HIV RRE–Rev binding28, 29 and, more recently, monitoring the binding of aminoglycoside antibiotics to the bacterial decoding site30, 31 and HIV-1 dimerization initiation site32.In contrast to the wide application of 2AP as a purine mimetic, the utilization of isosteric and responsive fluorescent pyrimidine analogues has not been explored in great detail33, 34, 35, 36, 37. To remedy this general deficiency in responsive pyrimidines, we have initiated a search for analogues that fulfill the criteria outlined above. A family of responsive fluorescent nucleoside analogues with emission in the visible range has recently been realized by conjugating five-membered aromatic heterocycles to the 5-position on the pyrimidine core38. In particular, a furan-conjugated 2′-deoxyuridine was found to be very sensitive to changes in solvent polarity, being rather emissive in polar environments (φ = 0.03 in water) and weakly emissive (φ<0.005) in apolar environments39, 40. We supposed that such behavior could be utilized to explore RNA recognition events, as these processes typically involve structural and dynamic changes that lead to changes in the microenvironment of participating reporter nucleotides. Indeed, the corresponding furan-modified uridine ribonucleoside was found to possess favorable solvent-sensitivity features41. Transcription using the required nucleotide triphosphate was chosen as a method for incorporating the fluorescent nucleotide, because of its simple synthesis—the triphosphate is synthesized in two steps from commercially available precursors—and, effective incorporation—reasonably large quantities of RNA can be synthesized by transcription reactions with relatively small amounts of the modified triphosphate. This makes the method accessible to many laboratories that are not necessarily equipped for complex organic synthesis and solid-phase RNA synthesis. We demonstrated the utility of the responsive fluorescent nucleoside analogue in RNA recognition events by effectively incorporating its nucleoside triphosphate into RNA constructs using in vitro transcription reactions, and exploring RNA-antibiotics41 and RNA–protein interactions42.The protocol outlined below provides a step-by-step synthesis of the furan-modified ribonucleoside 2 and its triphosphate 3, a necessary substrate for in vitro transcription reactions (Fig. 1). Detailed procedures for the enzymatic incorporation of triphosphate 3 into an RNA strand and purification of a modified oligoribonucleotide are given (Fig. 2). Also, this protocol briefly provides procedures for examining RNA–antibiotics interactions by fluorescence spectroscopy.Synthesis of nucleoside 2: Steps 1–7, 30 min; Steps 8–11, 10 min; Step 12, 2 h; Step 13, 5 min; Step 14, 30 min; Step 15, 15 min; Step 16, 20 min (evaporation depends on the volume and equipment used); Steps 17–19, 20 min; Steps 20–22, 30 min; Step 23, 5 h (total ~10 h).Synthesis of nucleoside triphosphate 3: Steps 24–26, 12 h; Steps 27–30, 20 min; Step 31, 30 h; Step 32, 5 min; Steps 33–35, 25 min; Steps 36 and 37, 15 min; Step 38, 30 min (evaporation depends on the volume and equipment used); Steps 39–42, 45 min; Steps 43 and 44, 11 h; Step 45, 1 h; Steps 46 and 47, 17 h; Step 48, 1.5 h; Steps 49 and 50, ~14 h; Steps 51–53, 36 h (total ~125 h).Purification of DNA oligonucleotides 4 and 5: Steps 54–57, 4.5 h; Steps 58–63, 12 h; Steps 64–66, 25 min; Steps 67–69, 40 min; Step 70, 6 h (time varies depending on the volume and instrument used); Step 71, 10 min (total ~24 h).Incorporation and purification of RNA containing nucleoside 2: Steps 72–74, 15 min; Steps 75–77, 1.5 h; Steps 78 and 79, 25 min; Step 80, 12 h; Steps 81–83, 1.5 h; Steps 84 and 85, 5 min; Step 86, ~24 h (total ~40 h).Troubleshooting advice can be found in Table 1.A duplex A-site RNA construct was formed by heating 1:1 mixture of furan-modified ssRNA 6 and custom-synthesized complementary ssRNA 7 at 75 °C for 5 min in HEPES buffer (20 mM HEPES, 100 mM NaCl, 0.5 mM EDTA, pH 7.4) and cooling the solution slowly to room temperature, followed by incubating the mixture on ice (Fig. 4). Steady-state fluorescence spectra of the duplex were measured on a Perkin Elmer LS 50B luminescence spectrometer. Duplex RNA was excited at 322 nm with an excitation slit width of 15 nm and emission slit width of 20 nm, and changes in fluorescence upon aminoglycoside binding were monitored at the emission maximum, 433 nm (Fig. 4) (see ref. 41). Titration of paromomycin and neomycin to the duplex resulted in nearly twofold increase in emission intensity corresponding to an EC50 value of 11.5 ± 0.9 and 5.0 ± 0.24 μM, respectively.
1H-NMR (400 MHz, d
6-DMSO): δ 11.66 (s, 1H), 8.42 (s, 1H), 7.61 (s, 1H), 6.86 (d, J = 3.2 Hz, 1H), 6.53–6.52 (m, 1H), 5.86 (d, J = 5.2 Hz, H1′, 1H), 5.43 (d, J = 5.6 Hz, OH, 1H), 5.20 (t, J = 4.6 Hz, OH, 1H), 5.11 (d, J = 5.2 Hz, OH, 1H), 4.13–4.0 (m, H2′, H3′, 2H), 3.90 (d, J = 3.2 Hz, H4′, 1H), 3.71–3.57 (m, H5′, 2H); 13C-NMR (100 MHz, d
6-DMSO): δ 159.84, 149.44, 146.13, 141.34, 134.69, 111.42, 107.81, 105.51, 88.06, 84.82, 73.94, 69.80, 60.54; ESI-MS (m/z): calculated for C13H14N2O7 [M] 310.08, found [M + 1]+ = 310.84, [M + Na]+ = 332.94. λ
max (H2O) = 254 nm (ε
254 = 12,345 M−1 cm−1); λ
max (H2O) = 316 nm (ε
316 = 9,480 M−1 cm−1); ε
260 = 11,120 M−1 cm−1.
5-(fur-2-yl)uridine 5′-triphosphate (3)
1H-NMR (400 MHz, D2O): δ 8.25 (s, 1H), 7.63 (d, J = 0.8 Hz, 1H), 6.92 (d, J = 3.2 Hz, 1H), 6.57 (dd, J = 3.2 Hz, J = 1.6 Hz, 1H), 6.08 (d, J = 5.6 Hz, H1′, 1H), 4.54–4.48 (m, H2′, H3′, 2H), 4.36–4.28 (m, H4′, H5′, 3H), 3.02 (q, J = 7.2 Hz, (Et3NH+)4, 24H), 1.10 (t, J = 7.2 Hz, (Et3NH+)4, 36H); 13C-NMR (100 MHz, D2O): δ 162.34, 150.89, 145.23, 142.69, 135.50, 111.50, 108.98, 107.60, 88.52, 83.80 (d, J
PC = 9.2 Hz), 73.78, 70.02, 65.52 (d, J
PC = 7.2 Hz);31P-NMR (162 MHz, D2O): −9.19 (d, J = 20.4 Hz, Pγ), −10.78 (d, J = 20.3 Hz, Pα), −22.23 (t, J = 19.8 Hz, Pβ); ESI-MS (negative mode): calculated for C13H17N2O16P3 [M] 549.98, found [M−H]− = 548.92. The UV profile of the product is similar to that of the nucleoside 2.
Co-reporter:Victor K. Tam, Qi Liu and Yitzhak Tor
Chemical Communications 2006 (Issue 25) pp:2684-2686
Publication Date(Web):26 May 2006
DOI:10.1039/B604281C
Ethidium bromide has been extended by fusing an additional aromatic ring resulting in a larger intercalator with increased affinity for poly r(A)·r(U), poly d(A)·d(T) and triple helices when compared to the parent heterocycle.
Co-reporter:Nicholas J. Greco, Michelle Hysell, Joseph R. Goldenberg, Arnold L. Rheingold and Yitzhak Tor
Dalton Transactions 2006 (Issue 19) pp:2288-2290
Publication Date(Web):06 Apr 2006
DOI:10.1039/B603983A
A new alkyne-containing chelating ligand, 1,2-di(quinolin-8-yl)ethyne, is shown to form a mononuclear 1 : 1 complex with silver(I) and chelate the metal ion through both nitrogens and the ethyne moiety as seen by UV and NMR spectroscopy as well as X-ray crystallography.
Co-reporter:Kenneth F. Blount Dr. Dr.
ChemBioChem 2006 Volume 7(Issue 10) pp:
Publication Date(Web):17 AUG 2006
DOI:10.1002/cbic.200600109
Aminoglycoside antibiotics are RNA-binding polyamines that can bind with similar affinities to structurally diverse RNA targets. To design new semisynthetic aminoglycosides with improved target selectivity, it is important to understand the energetic and structural basis by which diverse RNA targets recognize similar ligands. It is also imperative to discover how novel aminoglycosides could be rationally designed to have enhanced selectivity for a given target. Two RNA drug targets, the prokaryotic ribosomal A-site and the HIV-1 TAR, provide an excellent model system in which to dissect the issue of target selectivity, in that they each have distinctive interactions with aminoglycosides. We report herein the design, synthesis, and binding activity of novel nucleobase–aminoglycoside conjugates that were engineered to be more selective for the A-site binding pocket. Contrary to the structural design, the conjugates bind the A-site more weakly than does the parent compound and bind the TAR more tightly than the parent compound. This result implies that the two RNA targets differ in their ability to adapt to structurally diverse ligands and thus have inherently different selectivities. This work emphasizes the importance of considering the inherent selectivity traits of the RNA target when engineering new ligands.
Co-reporter:Michelle Hysell, Jay S. Siegel and Yitzhak Tor
Organic & Biomolecular Chemistry 2005 vol. 3(Issue 16) pp:2946-2952
Publication Date(Web):19 Jul 2005
DOI:10.1039/B503757C
Synthesis and stability studies of exocyclic amino triazine nucleosides were performed. Stability of the nucleosides was found to be dependent on triazine ring electron density, solvent, and pH. The nucleosides were found to be more stable when the triazine ring was electron deficient, in high pH aqueous solutions and in aprotic solvents.
Co-reporter:Jürgen Boer Dr.;Kenneth F. Blount Dr.;Nathan W. Luedtke;Lev Elson-Schwab Dr.
Angewandte Chemie International Edition 2005 Volume 44(Issue 6) pp:
Publication Date(Web):3 JAN 2005
DOI:10.1002/anie.200461182
Revving up the RNA response: Binding of the anticancer drug cisplatin (cis-Pt) to nucleic acids is believed to favor DNA over RNA. To “reverse” the nucleic acid selectivity of cis-Pt, amino- and guanidinoglycoside–PtII conjugates (1 and 2) have been synthesized and evaluated. The conjugates generate site-specific, RNA-selective, covalent cross-links to the HIV-1 Rev response element.
Co-reporter:Nathan W. Luedtke Dr.;Qi Liu Dr.
Chemistry - A European Journal 2005 Volume 11(Issue 2) pp:
Publication Date(Web):17 NOV 2004
DOI:10.1002/chem.200400559
The electronic structure of the common intercalating agent ethidium bromide (3,8-diamino-5-ethyl-6-phenylphenanthridinium bromide) is dominated by an interplay of electron donating and withdrawing effects mediated by its nitrogen atoms. X-ray crystallography, UV/Vis and IR absorption, fluorescence emission, and NMR spectroscopy are used to probe the electronic properties of the phenanthridinium “core” of ethidium as well as its exocyclic amines and 6-phenyl groups. Interestingly, despite its positive charge, most of ethidium's aromatic carbon and hydrogen atoms have high electron densities (compared to both 6-phenylphenanthridine and benzene). The data suggest that electron donation by ethidium's exocyclic amines dominates over the electron withdrawing effects of its endocyclic iminium in their combined influence on the electron densities of these atoms. Ethidium's nitrogen atoms are, conversely, electron deficient where the 5-position is the most electropositive, followed by the 3-amino, and lastly the 8-amino group. These results have been used to generate an empirically-based π-electron density map of ethidium that may prove useful to understanding its nucleic acid binding specificity.
Co-reporter:Jürgen Boer Dr.;Kenneth F. Blount Dr.;Nathan W. Luedtke;Lev Elson-Schwab Dr.
Angewandte Chemie 2005 Volume 117(Issue 6) pp:
Publication Date(Web):3 JAN 2005
DOI:10.1002/ange.200461182
RNA bevorzugt: Das Tumortherapeutikum Cisplatin bindet stärker an DNA als an RNA. Um diese Selektivität für Nucleinsäuren umzukehren, wurden die PtII-Konjugate 1 und 2 mit einem Amino- bzw. Guanidinoglycosid synthetisiert und getestet. Die Konjugate erzeugen ortsspezifisch und RNA-selektiv kovalente Verknüpfungen zum HIV-1-Rev-Response-Element.
Co-reporter:Yitzhak Tor Dr.
ChemBioChem 2003 Volume 4(Issue 10) pp:
Publication Date(Web):26 SEP 2003
DOI:10.1002/cbic.200390111
The cover picture shows the processes involved in the search for small molecules as potent and selective RNA binders. Motivation comes from the desire to control cell function at the RNA level and to identify novel approaches to specifically combat pathogens by targeting their unique RNA sequences or RNA–protein complexes. Inspiration comes from nature; in particular, from aminoglycosides, a family of naturally occurring antibiotics that has been shown to target the bacterial ribosome. The discovery process involves identifying RNA targets (schematically shown as a ribosome or a virus), devising unique assays (e.g. a solid-phase assay), and generating the necessary knowledge and lead structures through design, synthesis, and systematic evaluation of biological activity. Further details can be found in the article by Y. Tor on p. 998 ff.
Co-reporter:Yitzhak Tor Dr.
ChemBioChem 2003 Volume 4(Issue 10) pp:
Publication Date(Web):26 SEP 2003
DOI:10.1002/cbic.200300680
Revealing the “rules” that govern RNA–ligand recognition will have an impact on our ability to ultimately control cell function at the RNA level and combat pathogens by specifically targeting their RNA or RNA–protein complexes.
Co-reporter:Nathan W. Luedtke
Biopolymers 2003 Volume 70(Issue 1) pp:
Publication Date(Web):29 JUL 2003
DOI:10.1002/bip.10428
RNA plays a pivotal role in the replication of all organisms, including viral and bacterial pathogens. The development of small molecules that selectively interfere with undesired RNA activity is a promising new direction for drug design. Currently, there are no anti-HIV treatments that target nucleic acids. This article presents the HIV-1 Rev response element (RRE) as an important focus for the development of antiviral agents that target RNA. The Rev binding site on the RRE is highly conserved, even between different groups of HIV-1 isolates. Compounds that inhibit HIV replication by binding to the RRE and displacing Rev are therefore expected to retain activity across groups of genetically diverse HIV infections. Systematic evaluations of both the RRE affinity and specificity of numerous small molecule inhibitors are essential for deciphering the parameters that govern effective RRE recognition. This article discusses fluorescence-based techniques that are useful for probing a small molecule's RRE affinity and its ability to inhibit Rev–RRE binding. Rev displacement experiments can be conducted by observing the fluorescence anisotropy of a fluorescein-labeled Rev peptide, or by quantifying its displacement from a solid-phase immobilized RRE. Experiments conducted in the presence of competing nucleic acids are useful for evaluating the RRE specificity of Rev-RRE inhibitors. The discovery and characterization of new RRE ligands are described. Eilatin is a polycyclic aromatic heterocycle that has at least one binding site on the RRE (apparent Kd ≈ 0.13 μM), but it does not displace Rev upon binding the RRE (IC50 > 3 μM). In contrast, ethidium bromide and two eilatin-containing metal complexes show better consistency between their RRE affinity and their ability to displace a fluorescent Rev peptide from the RRE. These results highlight the importance of conducting orthogonal binding assays that establish both the RNA affinity of a small molecule and its ability to inhibit the function of the RNA target. Some Rev–RRE inhibitors, including ethidium bromide, Λ-[Ru(bpy)2eilatin]2+, and Δ-[Ru(bpy)2eilatin]2+ also inhibit HIV-1 gene expression in cell cultures (IC50 = 0.2–3 μM). These (and similar) results should facilitate the future discovery and implementation of anti-HIV drugs that are targeted to viral RNA sites. In addition, a deeper general understanding of RNA–small molecule recognition will assist in the effective targeting of other therapeutically important RNA sites. © 2003 Wiley Periodicals, Inc. Biopolymers, 2003
Co-reporter:Edith C. Glazer Dr.
Angewandte Chemie International Edition 2002 Volume 41(Issue 21) pp:
Publication Date(Web):31 OCT 2002
DOI:10.1002/1521-3773(20021104)41:21<4022::AID-ANIE4022>3.0.CO;2-1
The fabrication of RuII–polypyridyl complexes with “large-surface” ligands can be achieved by utilizing metal coordination to facilitate a dehydrogenation reaction (see scheme). This modular methodology efficiently yields biologically active eilatin-containing complexes, as well as RuII complexes of previously unknown ligands.
Co-reporter:Nathan W. Luedtke;Judy S. Hwang;Edith C. Glazer;Dalia Gut;Moshe Kol Dr‥
ChemBioChem 2002 Volume 3(Issue 8) pp:
Publication Date(Web):29 JUL 2002
DOI:10.1002/1439-7633(20020802)3:8<766::AID-CBIC766>3.0.CO;2-X
Eilatin-containing octahedral ruthenium complexes inhibit HIV-1 replication in CD4+ HeLa cells and in human peripheral blood monocytes with IC50values of approximately 1 μM. Similar metal complexes that lack eilatin display 15–100-fold lower anti-HIV activities. [Ru(bpy)2“pre-eilatin”]2+, a complex that contains a nonplanar analogue of eilatin, shows significantly lower nucleic acid binding and lower anti-HIV activity than eilatin complexes. This result indicates that the extended planar surface presented by eilatin is important for both activities. Rev peptide and ethidium bromide displacement assays are used to probe the nucleic acid affinity and specificity of Λ- and Δ-[Ru(bpy)2eilatin]2+. Two HIV-1 RNA sites are compared and a significant binding preference for the Rev response element over the transactivation response region is found. Simple DNA duplexes show a consistent selectivity for Λ-[Ru(bpy)2eilatin]2+compared to Δ-[Ru(bpy)2eilatin]2+, while RNAs show more diverse enantiomeric selectivities.
Co-reporter:Edith C. Glazer Dr.
Angewandte Chemie 2002 Volume 114(Issue 21) pp:
Publication Date(Web):31 OCT 2002
DOI:10.1002/1521-3757(20021104)114:21<4194::AID-ANGE4194>3.0.CO;2-M
Platt durch Wasserstoffentzug: Eine Dehydrierung am metallkoordinierten Liganden ist der Schlüsselschritt beim Aufbau von RuII-Polypyridylkomplexen mit großen und flachen Liganden (siehe Schema). Durch die modulare Bauweise lassen sich nicht nur die biologisch aktiven RuII-Eilatin-Komplexe, sondern auch Komplexe mit bislang unbekannten Liganden herstellen.
Co-reporter:Hima S. Joshi and Yitzhak Tor
Chemical Communications 2001 (Issue 6) pp:549-550
Publication Date(Web):28 Feb 2001
DOI:10.1039/B100036P
A RuII–based emission, while almost entirely
quenched in a RuII/OsII heterodimetallated DNA
hairpin, is dramatically restored upon hybridization to a complementary
oligonucleotide, while hybridization to an oligonucleotide that contains a
single mismatch results in significantly lower emission intensity.
Co-reporter:Haim Weizman and Yitzhak Tor
Chemical Communications 2001 (Issue 5) pp:453-454
Publication Date(Web):14 Feb 2001
DOI:10.1039/B009855H
A novel ‘ligandoside’ dimer, where
2,2′-bipyridine chelators are attached to the anomeric carbons of a
D-ribose–phosphate backbone, forms double stranded
multinuclear complexes.
Co-reporter:Nathan W. Luedtke Dr.
Angewandte Chemie 2000 Volume 112(Issue 10) pp:
Publication Date(Web):15 MAY 2000
DOI:10.1002/(SICI)1521-3757(20000515)112:10<1858::AID-ANGE1858>3.0.CO;2-8
Co-reporter:Hima S. Joshi;Ramin Jamshidi
Angewandte Chemie 1999 Volume 111(Issue 18) pp:
Publication Date(Web):15 SEP 1999
DOI:10.1002/(SICI)1521-3757(19990917)111:18<2887::AID-ANGE2887>3.0.CO;2-W
Mehrere Emissionsfarben können mit derselben Verbindung aus einer neuartigen Klasse stark lichtemittierender und im sichtbaren Spektralbereich fluoreszierender 1,10-Phenanthroline 1 erzeugt werden. Die Emissionswellenlänge jedes einzelnen Derivates wird durch die Natur seiner Substituenten bestimmt und kann durch die Zugabe exogener Additive wie Protonen oder Metallionen weiter moduliert werden. R=H, Me, OMe, NMe2.
Co-reporter:Yitzhak Tor
Angewandte Chemie 1999 Volume 111(Issue 11) pp:
Publication Date(Web):26 MAY 1999
DOI:10.1002/(SICI)1521-3757(19990601)111:11<1681::AID-ANGE1681>3.0.CO;2-6
Eine Schlüsselrolle bei lebenswichtigen zellulären Prozessen spielen RNA-Moleküle, die deshalb interessante Ziele für das Wirkstoff-Design sind. Die Vielfältigkeit der Funktionen von RNA kann auf die komplexen dreidimensionalen Strukturen zurückgeführt werden, die diese einnehmen kann. Die komplizierten Faltungsmuster enthalten potentielle Bindungstaschen für Ionen, niedermolekulare Liganden und Proteine. Neuere Untersuchungen haben ergeben, daß kleine Moleküle wie Tobramycin 1 die Genexpression in lebenden Zellen durch spezifische Wechselwirkungen mit einer Messenger-RNA (mRNA) regulieren können.
Co-reporter:Hima S. Joshi;Ramin Jamshidi
Angewandte Chemie International Edition 1999 Volume 38(Issue 18) pp:
Publication Date(Web):15 SEP 1999
DOI:10.1002/(SICI)1521-3773(19990917)38:18<2721::AID-ANIE2721>3.0.CO;2-5
Multiple emission colors can be generated with the same compound from a novel family of highly emissive and visibly fluorescent 1,10-phenanthrolines 1. The emission wavelength of any derivative is dictated by the nature of its substituent and can be further modulated by exogenous additives such as protons or metal ions. R=H, Me, OMe, NMe2.
Co-reporter:Yitzhak Tor
Angewandte Chemie International Edition 1999 Volume 38(Issue 11) pp:
Publication Date(Web):26 MAY 1999
DOI:10.1002/(SICI)1521-3773(19990601)38:11<1579::AID-ANIE1579>3.0.CO;2-H
A key role in essential cellular processes is played by RNA molecules, and these are attractive targets for drug design. The functional diversity of RNA can be attributed to the sophisticated three-dimensional structures it assumes. These intricate folds create potential binding pockets for ions, low molecular weight ligands, and proteins. Recent experiments have demonstrated that small molecules such as tobramycin (1) can regulate gene expression in living cells through specific interactions with a messenger RNA (mRNA).
Co-reporter:Hai Wang
Angewandte Chemie International Edition 1998 Volume 37(Issue 1‐2) pp:
Publication Date(Web):17 DEC 1998
DOI:10.1002/(SICI)1521-3773(19980202)37:1/2<109::AID-ANIE109>3.0.CO;2-0
The critical role of electrostatic interactions in the binding of aminoglycoside antibiotics to RNA is demonstrated by replacing the OH group, for example in kanamycin A (1), by NH2. Derivative 2 inhibits a hammerhead ribosyme much more strongly than 1. It is proposed that the three-dimensional arrangement of the protonated NH2 groups determines the binding strength of the aminoglycosides.
Co-reporter:Hai Wang
Angewandte Chemie 1998 Volume 110(Issue 1‐2) pp:
Publication Date(Web):12 MAR 1999
DOI:10.1002/(SICI)1521-3757(19980116)110:1/2<117::AID-ANGE117>3.0.CO;2-#
Die elektrostatische Wechselwirkung ist entscheidend bei der Bindung von Aminoglycosid-Antibiotica an RNA. Dies ergab der Austausch von OH- gegen NH2-Gruppen, z.B. in Kanamycin A 1. Das Derivat 2 inhibiert ein Hammerhead-Ribozym viel stärker als 1. Es wird vorgeschlagen, daß die räumliche Anordnung der positiven Ladungen die Bindungsstärke der Aminoglycoside bestimmt.
Co-reporter:Katja Michael
Chemistry - A European Journal 1998 Volume 4(Issue 11) pp:
Publication Date(Web):14 DEC 1998
DOI:10.1002/(SICI)1521-3765(19981102)4:11<2091::AID-CHEM2091>3.0.CO;2-3
The successful design of specific RNA binders requires intimate knowledge of RNA structure, folding, and recognition. Investigation of the interactions between aminoglycoside antibiotics (such as kanamycin A and B, R1 = OH and NH2, respectively, R2 = OH) and RNA has unraveled certain recognition rules; we propose a recognition model suggesting three-dimensional electrostatic complementarity and discuss issues of binding specificity and factors that have to be considered in designing new RNA binders.
Co-reporter:Yun Xie ; Andrew V. Dix
Journal of the American Chemical Society () pp:
Publication Date(Web):November 12, 2009
DOI:10.1021/ja905767g
A robust analysis and discovery platform for antibiotics targeting the bacterial rRNA A-site has been developed by incorporating a new emissive U surrogate into the RNA and labeling the aminoglycosides with an appropriate fluorescence acceptor. Specifically, a 5-methoxyquinazoline-2,4(1H,3H)-dione-based emissive uracil analogue was identified to be an ideal donor for 7-diethylaminocoumarin-3-carboxylic acid. This donor/acceptor pair displays a critical Förster radius (R0) of 27 Å, a value suitable for an A-site-aminoglycoside assembly. Titrating the coumarin labeled aminoglycosides into the emissive A-site construct, labeled at position U1406, shows a decrease in donor emission (at 395 nm) and concurrent increase of the acceptor emission (at 473 nm). Titration curves, obtained by fitting the donor’s emission quenching or the augmentation of the acceptor’s sensitized emission, faithfully generate EC50 values. Titration of unlabeled ligands into the preformed FRET complex showed a continuous increase of the donor emission, with a concurrent decrease of the acceptor emission, yielding valuable data regarding competitive displacement of aminoglycosides by A-site binders. Detection of antibiotic binding is therefore not dependent on changes in the environment of a single fluorophore, but rather on the responsive interaction between two chromophores acting as a FRET pair, facilitating the determination of direct binding and competitive displacement events with FRET accuracy.
Co-reporter:Marianna Sholokh; Rajhans Sharma; Dongwon Shin; Ranjan Das; Olga A. Zaporozhets; Yitzhak Tor;Yves Mély
Journal of the American Chemical Society () pp:
Publication Date(Web):February 25, 2015
DOI:10.1021/ja513107r
The archetypical fluorescent nucleoside analog, 2-aminopurine (2Ap), has been used in countless assays, though it suffers from very low quantum yield, especially when included in double strands, and from the fact that its residual emission frequently does not represent biologically relevant conformations. To conquer 2Ap’s deficiencies, deoxythienoguanosine (dthG) was recently developed. Here, steady-state and time-resolved fluorescence spectroscopy was used to compare the ability of 2Ap and dthG, to substitute and provide relevant structural and dynamical information on a key G residue in the (−) DNA copy of the HIV-1 primer binding site, (−)PBS, both in its stem loop conformation and in the corresponding (−)/(+)PBS duplex. In contrast to 2Ap, this fluorescent nucleoside when included in (−)PBS or (−)/(+)PBS duplex fully preserves their stability and exhibits a respectable quantum yield and a simple fluorescence decay, with marginal amounts of dark species. In further contrast to 2Ap, the fluorescently detected dthG species reflect the predominantly populated G conformers, which allows exploring their relevant dynamics. Being able to perfectly substitute G residues, dthG will transform nucleic acid biophysics by allowing, for the first time, to selectively and faithfully monitor the conformations and dynamics of a given G residue in a DNA sequence.
Co-reporter:Kristina M. Hamill, Ezequiel Wexselblatt, Wenyong Tong, Jeffrey D. Esko and Yitzhak Tor
Journal of Materials Chemistry A 2016 - vol. 4(Issue 35) pp:NaN5797-5797
Publication Date(Web):2016/08/09
DOI:10.1039/C6TB01387B
Two methods for assembling guanidinoneomycin-decorated liposomes are presented and their ability to deliver an active enzyme to the lysosomes and restore enzyme function in diseased cells is compared.
Co-reporter:Kristina M. Hamill, Lisa S. McCoy, Ezequiel Wexselblatt, Jeffrey D. Esko and Yitzhak Tor
Chemical Science (2010-Present) 2016 - vol. 7(Issue 8) pp:
Publication Date(Web):
DOI:10.1039/C6SC00488A
Co-reporter:C. Vranken, A. Fin, P. Tufar, J. Hofkens, M. D. Burkart and Y. Tor
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 26) pp:NaN6192-6192
Publication Date(Web):2016/05/24
DOI:10.1039/C6OB00844E
SalL, an enzyme that catalyzes the synthesis of SAM from L-methionine and 5′-chloro-5′-deoxyoadenosine, is shown to accept 5′-chloro-5′-deoxythienoadenosine as a substrate and facilitate the synthesis of a synthetic SAM analog with an unnatural nucleobase. This synthetic cofactor is demonstrated to replace SAM in the DNA methylation reaction with M.TaqI.
Co-reporter:Seergazhi G. Srivatsan, Haim Weizman and Yitzhak Tor
Organic & Biomolecular Chemistry 2008 - vol. 6(Issue 8) pp:NaN1338-1338
Publication Date(Web):2008/03/10
DOI:10.1039/B801054D
A highly emissive nucleobase analog, based on a thieno[3,4-d]pyrimidine core, is enzymatically incorporated into RNA oilgonucleotides that function as base discriminating fluorescent probes.
Co-reporter:Matthew J. Belousoff, Bim Graham, Leone Spiccia and Yitzhak Tor
Organic & Biomolecular Chemistry 2009 - vol. 7(Issue 1) pp:NaN33-33
Publication Date(Web):2008/10/30
DOI:10.1039/B813252F
A number of aminoglycoside antibiotics, and in particular neomycin B, are demonstrated to promote strand cleavage of RNA oligonucleotides (minimised HIV-1 TAR element and prokaryotic ribosomal A-site), by binding and causing sufficient distortion to the RNA backbone to render it more susceptible to intramolecular transesterification.
Co-reporter:Yun Xie, Tucker Maxson and Yitzhak Tor
Organic & Biomolecular Chemistry 2010 - vol. 8(Issue 22) pp:NaN5055-5055
Publication Date(Web):2010/09/23
DOI:10.1039/C0OB00413H
A fluorescent nucleobase analogue, 7-aminoquinazoline-2,4-(1H,3H)-dione, is incorporated into a DNA oligonucleotide and senses mismatched pairing by displaying G-specific fluorescence enhancement.
Co-reporter:Ryan J. Weiss, Philip L. S. M. Gordts, Dzung Le, Ding Xu, Jeffrey D. Esko and Yitzhak Tor
Chemical Science (2010-Present) 2015 - vol. 6(Issue 10) pp:NaN5993-5993
Publication Date(Web):2015/07/29
DOI:10.1039/C5SC01208B
Surfen, bis-2-methyl-4-amino-quinolyl-6-carbamide, was previously reported as a small molecule antagonist of heparan sulfate (HS), a key cell-surface glycosaminoglycan found on all mammalian cells. To generate structure–activity relationships, a series of rationally designed surfen analogs was synthesized, where its dimeric structure, exocyclic amines, and urea linker region were modified to probe the role of each moiety in recognizing HS. An in vitro assay monitoring inhibition of fibroblast growth factor 2 binding to wild-type CHO cells was utilized to quantify interactions with cell surface HS. The dimeric molecular structure of surfen and its aminoquinoline ring systems was essential for its interaction with HS, and certain dimeric analogs displayed higher inhibitory potency than surfen and were also shown to block downstream FGF signaling in mouse embryonic fibroblast cells. These molecules were also able to antagonize other HS–protein interactions including the binding of soluble RAGE to HS. Importantly, selected molecules were shown to neutralize heparin and other heparinoids, including the synthetic pentasaccharide fondaparinux, in a factor Xa chromogenic assay and in vivo in mice. These results suggest that small molecule antagonists of heparan sulfate and heparin can be of therapeutic potential for the treatment of disorders involving glycosaminoglycan–protein interactions.
Co-reporter:Yun Xie, Andrew V. Dix and Yitzhak Tor
Chemical Communications 2010 - vol. 46(Issue 30) pp:NaN5544-5544
Publication Date(Web):2010/05/13
DOI:10.1039/C0CC00423E
A FRET assembly reports antibiotic affinities to two different RNA targets. A binder was labeled with a fluorophore that acts both as an acceptor for the emissive nucleoside on the bacterial A-site and a donor fluorophore for the terminally-labeled human A-site. Unlabeled drugs were used to dissociate the labeled antibiotic.
Co-reporter:Dongwon Shin, Peter Lönn, Steven F. Dowdy and Yitzhak Tor
Chemical Communications 2015 - vol. 51(Issue 9) pp:NaN1665-1665
Publication Date(Web):2014/12/05
DOI:10.1039/C4CC08809C
Singly and multiply modified synthetic siRNA oligonucleotides, containing isomorphic surrogate nucleobases, show high interference potency in cell culture, suggesting the highly isomorphic RNA alphabet, based on a thieno[3,4-d]-pyrimidine core, is tolerated well by the cellular silencing machinery.
Co-reporter:Alexander R. Rovira, Andrea Fin and Yitzhak Tor
Chemical Science (2010-Present) 2017 - vol. 8(Issue 4) pp:NaN2993-2993
Publication Date(Web):2017/01/30
DOI:10.1039/C6SC05354H
A series of emissive ribonucleoside purine mimics, all comprised of an isothiazolo[4,3-d]pyrimidine core, was prepared using a divergent pathway involving a key Thorpe–Ziegler cyclization. In addition to an adenosine and a guanosine mimic, analogues of the noncanonical xanthosine, isoguanosine, and 2-aminoadenosine were also synthesized and found to be emissive. Isothiazolo 2-aminoadenosine, an adenosine surrogate, was found to be particularly emissive and effectively deaminated by adenosine deaminase. Competitive studies with adenosine deaminase with each analogue in combination with native adenosine showed preference for the native substrate while still deaminating the isothiazolo analogues.
Co-reporter:Ryan J. Weiss, Jeffrey D. Esko and Yitzhak Tor
Organic & Biomolecular Chemistry 2017 - vol. 15(Issue 27) pp:NaN5668-5668
Publication Date(Web):2017/06/13
DOI:10.1039/C7OB01058C
Heparin and heparan sulfate glycosaminoglycans are long, linear polysaccharides that are made up of alternating dissacharide sequences of sulfated uronic acid and amino sugars. Unlike heparin, which is only found in mast cells, heparan sulfate is ubiquitously expressed on the cell surface and in the extracellular matrix of all animal cells. These negatively-charged glycans play essential roles in important cellular functions such as cell growth, adhesion, angiogenesis, and blood coagulation. These biomolecules are also involved in pathophysiological conditions such as pathogen infection and human disease. This review discusses past and current methods for targeting these complex biomolecules as a novel therapeutic strategy to treating disorders such as cancer, neurodegenerative diseases, and infection.
Co-reporter:Patrycja A. Hopkins, Lisa S. McCoy and Yitzhak Tor
Organic & Biomolecular Chemistry 2017 - vol. 15(Issue 3) pp:NaN690-690
Publication Date(Web):2016/12/12
DOI:10.1039/C6OB02080A
To display favorable fluorescent properties, the non-emissive native nucleosides need to be modified. Here we present a motif that relies on conjugating 5-membered aromatic heterocycles (e.g., thiophene) to a 6-azapyrimidine (1,2,4-triazine) core. Synthetic accessibility and desirable photophysical properties make these nucleosides attractive candidates for enzymatic incorporation and biochemical assays. While 6-azauridine triphosphate is known to be poorly tolerated by polymerases in RNA synthesis, we illustrate that conjugating a thiophene ring at position 5 overcomes such limitations, facilitating its T7 RNA polymerase-mediated in vitro transcription incorporation into RNA constructs. We further show that the modified transcripts can be ligated to longer oligonucleotides to form singly modified RNAs, as illustrated for an A-site hairpin model RNA construct, which was employed to visualize aminoglycoside antibiotics binding.
.jpg)
![1,2,4-Triazine-3,5(2H,4H)-dione,2-[5-O-[hydroxy[[hydroxy(phosphonooxy)phosphinyl]oxy]phosphinyl]-b-D-ribofuranosyl]-](http://img.cochemist.com/ccimg/6200/6198-30-7.png)
![1,2,4-Triazine-3,5(2H,4H)-dione,2-[5-O-[hydroxy[[hydroxy(phosphonooxy)phosphinyl]oxy]phosphinyl]-b-D-ribofuranosyl]-](http://img.cochemist.com/ccimg/6200/6198-30-7_b.png)
![Butanoic acid, 4-[[2-[2-[2-(2-azidoethoxy)ethoxy]ethoxy]ethyl]amino]-4-oxo-](/data/chemimg/3722900/1202400-17-6.png)
![Butanoic acid, 4-[[2-[2-[2-(2-azidoethoxy)ethoxy]ethoxy]ethyl]amino]-4-oxo-](/data/chemimg/3722900/1202400-17-6_b.png)
![Ethanamine, 2-[2-[2-(2-azidoethoxy)ethoxy]ethoxy]-](/data/chemimg/114300/134179-38-7.png)
![Ethanamine, 2-[2-[2-(2-azidoethoxy)ethoxy]ethoxy]-](/data/chemimg/114300/134179-38-7_b.png)

![Octadecanamide, N-[2-[2-[2-(2-propyn-1-yloxy)ethoxy]ethoxy]ethyl]-](/data/chemimg/3723500/1703784-45-5.png)
![Octadecanamide, N-[2-[2-[2-(2-propyn-1-yloxy)ethoxy]ethoxy]ethyl]-](/data/chemimg/3723500/1703784-45-5_b.png)
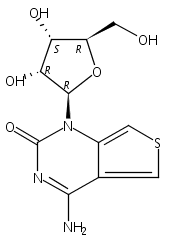
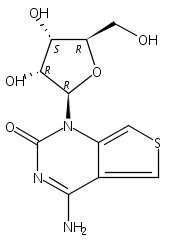
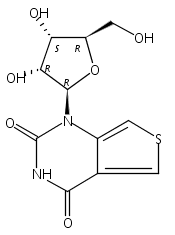
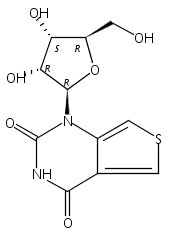


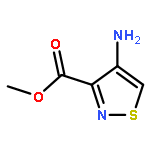
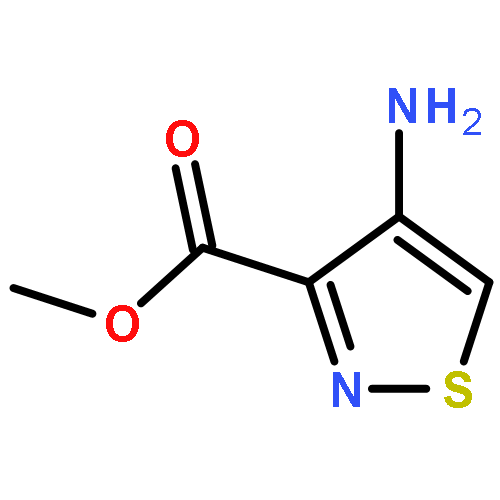
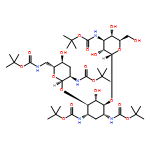
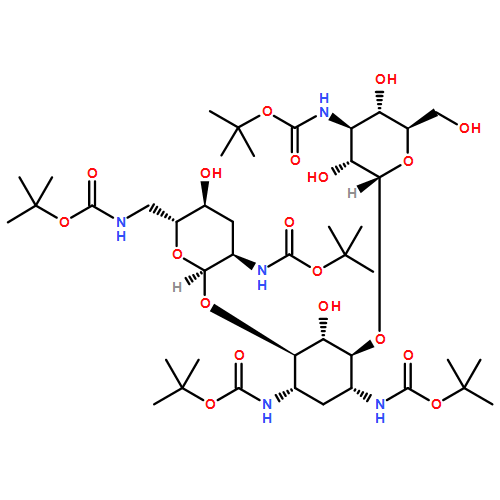


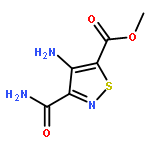
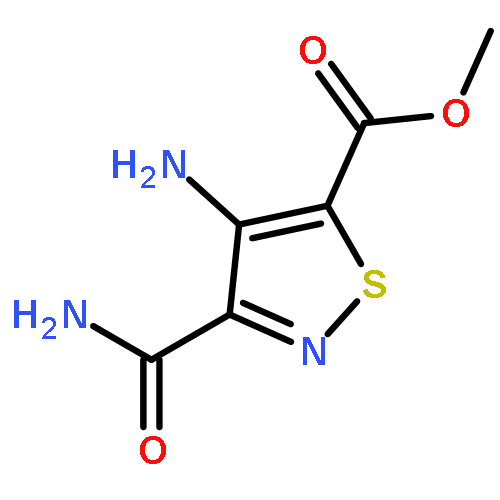
![1-Propanaminium,N,N,N-trimethyl-2,3-bis[[(9Z)-1-oxo-9-octadecen-1-yl]oxy]-](http://img.cochemist.com/ccimg/113700/113669-21-9.png)
![1-Propanaminium,N,N,N-trimethyl-2,3-bis[[(9Z)-1-oxo-9-octadecen-1-yl]oxy]-](http://img.cochemist.com/ccimg/113700/113669-21-9_b.png)
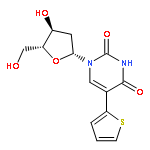

![[(2-AMINO-1-CYANO-2-OXOETHYLIDENE)AMINO] 4-METHYLBENZENESULFONATE](http://img.cochemist.com/ccimg/55000/54968-77-3.png)
![[(2-AMINO-1-CYANO-2-OXOETHYLIDENE)AMINO] 4-METHYLBENZENESULFONATE](http://img.cochemist.com/ccimg/55000/54968-77-3_b.png)

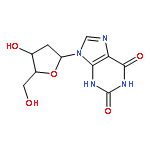
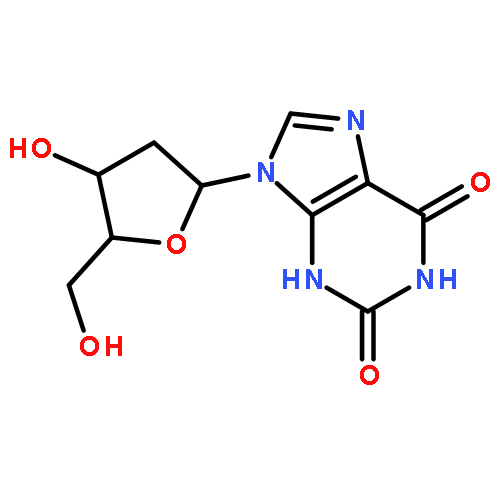
![3,5,8-Trioxa-4-phosphahexacos-17-en-1-aminium,4-hydroxy-N,N,N-trimethyl-9-oxo-7-[[(1-oxohexadecyl)oxy]methyl]-, inner salt,4-oxide, (7R,17Z)-](http://img.cochemist.com/ccimg/26900/26853-31-6.png)
![3,5,8-Trioxa-4-phosphahexacos-17-en-1-aminium,4-hydroxy-N,N,N-trimethyl-9-oxo-7-[[(1-oxohexadecyl)oxy]methyl]-, inner salt,4-oxide, (7R,17Z)-](http://img.cochemist.com/ccimg/26900/26853-31-6_b.png)

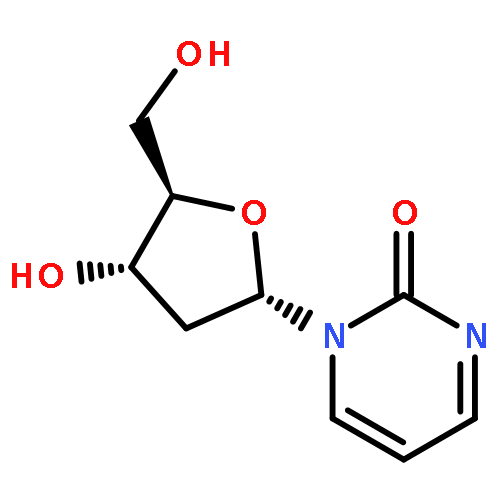




![3,5,9-Trioxa-4-phosphaheptacos-18-en-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[[(9Z)-1-oxo-9-octadecen-1-yl]oxy]-, innersalt, 4-oxide, (7R,18Z)-](http://img.cochemist.com/ccimg/4300/4235-95-4.png)
![3,5,9-Trioxa-4-phosphaheptacos-18-en-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[[(9Z)-1-oxo-9-octadecen-1-yl]oxy]-, innersalt, 4-oxide, (7R,18Z)-](http://img.cochemist.com/ccimg/4300/4235-95-4_b.png)
![9-Octadecenoic acid(9Z)-,1,1'-[(1R)-1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-1,2-ethanediyl]ester](http://img.cochemist.com/ccimg/4100/4004-05-1.png)
![9-Octadecenoic acid(9Z)-,1,1'-[(1R)-1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-1,2-ethanediyl]ester](http://img.cochemist.com/ccimg/4100/4004-05-1_b.png)