Co-reporter:Matthew D. Shortridge, Matthew J. Walker, Tom Pavelitz, Yu Chen, Wen Yang, and Gabriele Varani
ACS Chemical Biology June 16, 2017 Volume 12(Issue 6) pp:1611-1611
Publication Date(Web):April 24, 2017
DOI:10.1021/acschembio.7b00180
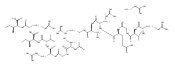
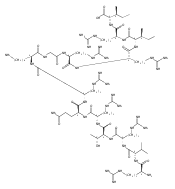
MicroRNAs (miRNAs) help orchestrate cellular growth and survival through post-transcriptional mechanisms. The dysregulation of miRNA biogenesis can lead to cellular growth defects and chemotherapeutic resistance and plays a direct role in the development of many chronic diseases. Among these RNAs, miR-21 is consistently overexpressed in most human cancers, leading to the down-regulation of key tumor-suppressing and pro-apoptotic factors, suggesting that inhibition of miR-21 biogenesis could reverse these negative effects. However, targeted inhibition of miR-21 using small molecules has had limited success. To overcome difficulties in targeting RNA secondary structure with small molecules, we developed a class of cyclic β-hairpin peptidomimetics which bind to RNA stem-loop structures, such as miRNA precursors, with potent affinity and specificity. We screened an existing cyclic peptide library and discovered a lead structure which binds to pre-miR21 with KD = 200 nM and prefers it over other pre-miRNAs. The NMR structure of the complex shows that the peptide recognizes the Dicer cleavage site and alters processing of the precursor to the mature miRNA in vitro and in cultured cells. The structure provides a rationale for the peptide binding activity and clear guidance for further improvements in affinity and targeting.
Co-reporter:Ravi P. Barnwal, Fan Yang, Gabriele Varani
Archives of Biochemistry and Biophysics 2017 Volume 628(Volume 628) pp:
Publication Date(Web):15 August 2017
DOI:10.1016/j.abb.2017.06.003
•Strategies for structure determination of RNA of different size.•NMR sample preparation, assignment strategies and structural constraints.•Hybrid techniques i.e., computational methods, cryo-EM, SAXS to supplement NMR.Nuclear magnetic resonance (NMR) spectroscopy is a powerful tool to investigate the structure and dynamics of RNA, because many biologically important RNAs have conformationally flexible structures, which makes them difficult to crystallize. Functional, independently folded RNA domains, range in size between simple stem-loops of as few as 10–20 nucleotides, to 50–70 nucleotides, the size of tRNA and many small ribozymes, to a few hundred nucleotides, the size of more complex RNA enzymes and of the functional domains of non-coding transcripts. In this review, we discuss new methods for sample preparation, assignment strategies and structure determination for independently folded RNA domains of up to 100 kDa in molecular weight.
Co-reporter:Francesco Musiani ; Giulia Rossetti ; Luciana Capece ; Thomas Martin Gerger ; Cristian Micheletti ; Gabriele Varani ;Paolo Carloni
Journal of the American Chemical Society 2014 Volume 136(Issue 44) pp:15631-15637
Publication Date(Web):October 14, 2014
DOI:10.1021/ja507812v
The HIV-1 Tat protein and several small molecules bind to HIV-1 transactivation responsive RNA (TAR) by selecting sparsely populated but pre-existing conformations. Thus, a complete characterization of TAR conformational ensemble and dynamics is crucial to understand this paradigmatic system and could facilitate the discovery of new antivirals targeting this essential regulatory element. We show here that molecular dynamics simulations can be effectively used toward this goal by bridging the gap between functionally relevant time scales that are inaccessible to current experimental techniques. Specifically, we have performed several independent microsecond long molecular simulations of TAR based on one of the most advanced force fields available for RNA, the parmbsc0 AMBER. Our simulations are first validated against available experimental data, yielding an excellent agreement with measured residual dipolar couplings and order parameter S2. This contrast with previous molecular dynamics simulations (Salmon et al., J. Am. Chem. Soc. 2013 135, 5457–5466) based on the CHARMM36 force field, which could achieve only modest accord with the experimental RDC values. Next, we direct the computation toward characterizing the internal dynamics of TAR over the microsecond time scale. We show that the conformational fluctuations observed over this previously elusive time scale have a strong functionally oriented character in that they are primed to sustain and assist ligand binding.
Co-reporter:Mi-Kyung Lee, Angel Bottini, Meehyein Kim, Michael F. Bardaro, Ziming Zhang, Maurizio Pellecchia, Byong-Seok Choi and Gabriele Varani
Chemical Communications 2014 vol. 50(Issue 83) pp:12578-12578
Publication Date(Web):12 Sep 2014
DOI:10.1039/C4CC90361G
Correction for ‘A novel small-molecule binds to the influenza A virus RNA promoter and inhibits viral replication’ by Mi-Kyung Lee et al., Chem. Commun., 2014, 50, 368–370.
Co-reporter:Mi-Kyung Lee, Angel Bottini, Meehyein Kim, Michael F. Bardaro, Ziming Zhang, Maurizio Pellecchia, Byong-Seok Choi and Gabriele Varani
Chemical Communications 2014 vol. 50(Issue 3) pp:368-370
Publication Date(Web):29 Oct 2013
DOI:10.1039/C3CC46973E
Through screening by NMR spectroscopy, we discovered a novel scaffold (DPQ: 6,7-dimethoxy-2-(1-piperazinyl)-4-quinazolinamine) that binds specifically to the influenza A virus RNA promoter. The solution structure of the RNA–DPQ complex reported here demonstrates that the internal loop is the binding site of DPQ. The scaffold exhibits antiviral activity against influenza viruses.
Co-reporter:Claire Moore;Susan D. Lee;Ravi Pratap Barnwal
PNAS 2012 Volume 109 (Issue 52 ) pp:21342-21347
Publication Date(Web):2012-12-26
DOI:10.1073/pnas.1214102110
The accuracy of the 3′-end processing by cleavage and polyadenylation is essential for mRNA biogenesis and transcription termination.
In yeast, two poorly conserved neighboring elements upstream of cleavage sites are important for accuracy and efficiency of
this process. These two RNA sequences are recognized by the RNA binding proteins Hrp1 and Rna15, but efficient processing
in vivo requires a bridging protein (Rna14), which forms a stable dimer of hetero-dimers with Rna15 to stabilize the RNA–protein
complex. We earlier reported the structure of the ternary complex of Rna15 and Hrp1 bound to the RNA processing element. We
now report the use of solution NMR to study the interaction of Hrp1 with the Rna14–Rna15 heterodimer in the presence and absence
of 3′-end processing signals. By using methyl selective labeling on Hrp1, in vivo activity and pull-down assays, we were able
to study this complex of several hundred kDa, identify the interface within Hrp1 responsible for recruitment of Rna14 and
validate the functional significance of this interaction through structure-driven mutational analysis.
Co-reporter:Yu Chen, Gabriele Varani
Chemistry & Biology 2011 Volume 18(Issue 7) pp:821-823
Publication Date(Web):29 July 2011
DOI:10.1016/j.chembiol.2011.07.001
Pumilio and FBF homology (PUF) proteins represent highly promising candidates for engineering sequence-specific RNA recognition, but were only known to recognize G, A, and U, significantly limiting applications. Two groups (Filipovska et al., 2011 and Dong et al., 2011) have now reported the discovery of the cytosine-recognition code for PUF proteins.
Co-reporter:Youhong Niu, Alisha “Jonesy” Jones, Haifan Wu, Gabriele Varani and Jianfeng Cai
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 19) pp:6604-6609
Publication Date(Web):01 Jul 2011
DOI:10.1039/C1OB05738C
The interactions between proteins and RNAs are of vital importance for many cellular processes, including transcription and processing of RNA, translation, and viral infections. Here we report an γ-AApeptide that can mimic HIV-1 Tat protein and bind to TAR RNAs of HIV and BIV with nanomolar affinity, comparable to that of the RNA-binding fragment of Tat (amino acids 49–58). The interaction is resistant to the presence of a large excess of tRNA. With resistance to proteolytic hydrolysis and limitless potential for diversification, γ-AApeptides may emerge as a new class of peptidomimetics to modulate RNA-protein interactions.
Co-reporter:Darren W. Begley;Suxin Zheng
Chemical Biology & Drug Design 2010 Volume 76( Issue 3) pp:218-233
Publication Date(Web):
DOI:10.1111/j.1747-0285.2010.01010.x
Solution-state nuclear magnetic resonance (NMR) is a versatile tool for the study of binding interactions between small molecules and macromolecular targets. We applied ligand-based NMR techniques to the study of human thymidylate synthase (hTS) using known nanomolar inhibitors and a library of small molecule fragments. Screening by NMR led to the rapid identification of ligand pairs that bind in proximal sites within the cofactor-binding pocket of hTS. Screening hits were used as search criteria within commercially available sources, and a subset of catalog analogs were tested for potency by in vitro assay and binding affinity by quantitative saturation transfer difference (STD)-NMR titration. Two compounds identified by this approach possess low micromolar affinity and potency, as well as excellent binding efficiency against hTS. Relative binding orientations for both leads were modeled using AutoDock, and the most likely bound conformations were validated using experimentally derived STD-NMR binding epitope data. These ligands represent novel starting points for fragment-based drug design of non-canonical TS inhibitors, and their binding epitopes highlight important and previously unexploited interactions with conserved residues in the cofactor-binding site.
Co-reporter:Mi-Kyung Lee;Maayan Gal;Lucio Frydman
PNAS 2010 Volume 107 (Issue 20 ) pp:9192-9197
Publication Date(Web):2010-05-18
DOI:10.1073/pnas.1001195107
Conformational transitions and structural rearrangements are central to the function of many RNAs yet remain poorly understood.
We have used ultrafast multidimensional NMR techniques to monitor the adenine-induced folding of an adenine-sensing riboswitch
in real time, with nucleotide-resolved resolution. By following changes in 2D spectra at rates of approximately 0.5 Hz, we
identify distinct steps associated with the ligand-induced folding of the riboswitch. Following recognition of the ligand,
long range loop-loop interactions form and are then progressively stabilized before the formation of a fully stable complex
over approximately 2–3 minutes. The application of these ultrafast multidimensional NMR methods provides the opportunity to
determine the structure of RNA folding intermediates and conformational trajectories.
Co-reporter:Suxin Zheng, Yu Chen, Christine P. Donahue, Michael S. Wolfe, Gabriele Varani
Chemistry & Biology 2009 Volume 16(Issue 5) pp:557-566
Publication Date(Web):29 May 2009
DOI:10.1016/j.chembiol.2009.03.009
Some familial neurodegenerative diseases are associated with mutations that destabilize a putative stem-loop structure within an intronic region of the tau pre-messenger RNA (mRNA) and alter the production of tau protein isoforms by alternative splicing. Because stabilization of the stem loop reverses the splicing pattern associated with neurodegeneration, small molecules that stabilize this stem loop would provide new ways to dissect the mechanism of neurodegeneration and treat tauopathies. The anticancer drug mitoxantrone was recently identified in a high throughput screen to stabilize the tau pre-mRNA stem loop. Here we report the solution structure of the tau mRNA-mitoxantrone complex, validated by the structure-activity relationship of existing mitoxantrone analogs. The structure describes the molecular basis for their interaction with RNA and provides a rational basis to optimize the activity of this new class of RNA-binding molecules.
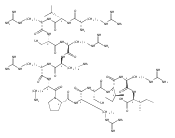
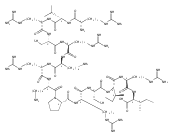
Co-reporter:Thomas C. Leeper;Krystyna Patora-Komisarska;Jonathan Karn;Amy Davidson;John A. Robinson;Zafiria Athanassiou
PNAS 2009 Volume 106 (Issue 29 ) pp:11931-11936
Publication Date(Web):2009-07-21
DOI:10.1073/pnas.0900629106
The interaction of the HIV-1 transactivator protein Tat with its transactivation response (TAR) RNA is an essential step in
viral replication and therefore an attractive target for developing antivirals with new mechanisms of action. Numerous compounds
that bind to the 3-nt bulge responsible for binding Tat have been identified in the past, but none of these molecules had
sufficient potency to warrant pharmaceutical development. We have discovered conformationally-constrained cyclic peptide mimetics
of Tat that are specific nM inhibitors of the Tat-TAR interaction by using a structure-based approach. The lead peptides are
nearly as active as the antiviral drug nevirapine against a variety of clinical isolates in human lymphocytes. The NMR structure
of a peptide–RNA complex reveals that these molecules interfere with the recruitment to TAR of both Tat and the essential
cellular cofactor transcription elongation factor-b (P-TEFb) by binding simultaneously at the RNA bulge and apical loop, forming
an unusually deep pocket. This structure illustrates additional principles in RNA recognition: RNA-binding molecules can achieve
specificity by interacting simultaneously with multiple secondary structure elements and by inducing the formation of deep
binding pockets in their targets. It also provides insight into the P-TEFb binding site and a rational basis for optimizing
the promising antiviral activity observed for these cyclic peptides.
Co-reporter:Steve L. Reichow and Gabriele Varani
Biochemistry 2008 Volume 47(Issue 23) pp:
Publication Date(Web):May 13, 2008
DOI:10.1021/bi800418p
The H/ACA class of small nucleolar ribonucleoproteins (snoRNPs) is primarily responsible for catalyzing the isomerization of uridine to pseudouridine (Ψ) in ribosomal and other cellular RNAs. Each H/ACA snoRNP consist of four conserved proteins, Cbf5 (the Ψ-synthase), Gar1, Nhp2 (L7Ae in archaea) and Nop10, that assemble onto a unique RNA component (the snoRNA). The smallest of these proteins, Nop10 (∼7 kDa), has an essential role in the assembly and activity of these particles and binds directly to the Ψ-synthase to form the minimal active enzyme in archaea. To better understand the conserved function of this protein, we characterized the NMR structure and dynamics of Nop10 proteins from both archaea and yeast. We show that archaeal Nop10 contains a highly stable Zn2+ binding motif that is replaced in eukaryotes by a smaller meta-stable β-hairpin, while a highly conserved and conformationally dynamic linker connects these motifs to a nascent α-helical structure. Our structural analysis and NMR relaxation data show that these motifs do not interact with each other and tumble independently in solution. Several residues within the archaeal Nop10 Zn2+ binding motif have clear structural and functional roles and are conserved in eukaryotes, yet remain disordered in the free yeast Nop10. We propose that the dynamic structure of Nop10 facilitates an induced-fit recognition with the H/ACA Ψ-synthase and allows it to act as a molecular adaptor for guiding snoRNP assembly in similar fashion in all archaea and eukaryotic organisms.
Co-reporter:Jiyun Liu;Darren Begley;Daniel D. Mitchell;Christophe L. M. J. Verlinde;Erkang Fan
Chemical Biology & Drug Design 2008 Volume 71( Issue 5) pp:408-419
Publication Date(Web):
DOI:10.1111/j.1747-0285.2008.00648.x
Multivalent inhibitors of the cholera toxin B pentamer are potential therapeutic drugs for treating cholera and serve as models for demonstrating multivalent ligand effects through a structure-based approach. A crucial yet often overlooked aspect of multivalent drug design is the length, rigidity and chemical composition of the linker used to connect multiple binding moieties. To specifically study the role of chemical linkers in multivalent ligand design, we have synthesized a series of compounds with one and two binding motifs connected by several different linkers. These compounds have affinity for and potency against the cholera toxin B pentamer despite the fact that none can simultaneously bind two toxin receptor sites. Results from saturation transfer difference NMR reveal transient, non-specific interactions between the cholera toxin and linker groups contribute significantly to overall binding affinity of monovalent compounds. However, the same random protein–ligand interactions do not appear to affect binding of bivalent molecules. Moreover, the binding affinities and potencies of these ‘non-spanning’ bivalent ligands appear to be wholly independent of linker length. Our detailed analysis identifies multiple effects that account for the improved inhibitory potencies of bivalent ligands and suggest approaches to further improve the activity of this class of compounds.
Co-reporter:Zahra Shajani and Gabriele Varani
Biochemistry 2008 Volume 47(Issue 29) pp:
Publication Date(Web):June 26, 2008
DOI:10.1021/bi7020469
The goal of this work was to examine if sequence-dependent conformational flexibility in DNA plays a role in base extrusion, a common conformational change induced by many DNA-modifying enzymes. We studied the dynamics of the double-stranded DNA target of the HhaI methyltransferase by recording an extensive set of 13C NMR relaxation parameters. We observe that the cytidine furanose rings experience fast (picosecond to nanosecond) motions that are not present in other nucleotides; the methylation site experiences particularly high mobility. We also observe that the bases of guanosine and cytidine residues within the HhaI recognition sequence GCGC experience motions on a much slower (1−100 μs) time scale. We compare these observations with previous solution and solid-state NMR studies of the EcoRI nuclease target sequence, and solid-state NMR studies of a similar HhaI target construct. While an increased mobility of cytidine furanose rings compared to those of other nucleotides is observed for both sequences, the slower motions are only observed in the HhaI target DNA. We propose that this inherent flexibility lowers the energetic barriers that must occur when the DNA binds to the HhaI methyltransferase and for extrusion of the cytidine prior to its methylation.
Co-reporter:Bradley M. Lunde,
Claire Moore
&
Gabriele Varani
Nature Reviews Molecular Cell Biology 2007 8(6) pp:479
Publication Date(Web):2007-06-01
DOI:10.1038/nrm2178
Many RNA-binding proteins have modular structures and are composed of multiple repeats of just a few basic domains that are arranged in various ways to satisfy their diverse functional requirements. Recent studies have investigated how different modules cooperate in regulating the RNA-binding specificity and the biological activity of these proteins. They have also investigated how multiple modules cooperate with enzymatic domains to regulate the catalytic activity of enzymes that act on RNA. These studies have shown how, for many RNA-binding proteins, multiple modules define the fundamental structural unit that is responsible for biological function.
Co-reporter:Zahra Shajani
Biopolymers 2007 Volume 86(Issue 5-6) pp:
Publication Date(Web):8 DEC 2006
DOI:10.1002/bip.20650
RNA and DNA molecules experience motions on a wide range of time scales, ranging from rapid localized motions to much slower collective motions of entire helical domains. The many functions of RNA in biology very often require this molecule to change its conformation in response to biological signals in the form of small molecules, proteins or other nucleic acids, whereas local motions in DNA may facilitate protein recognition and allow enzymes acting on DNA to access functional groups on the bases that would otherwise be buried in Watson-Crick base pairs. Although these statements make a compelling case to study the sequence dependent dynamics in nucleic acids, there are few residue-specific studies of nucleic acid dynamics. Fortunately, NMR studies of dynamics of nucleic acids and nucleic acids-protein complexes are gaining increased attention. The aim of this review is to provide an update of the recent progress in studies of nucleic acid dynamics by NMR based on the application of solution relaxation techniques. © 2006 Wiley Periodicals, Inc. Biopolymers 86: 348–359, 2007.
This article was originally published online as an accepted preprint. The “Published Online” date corresponds to the preprint version. You can request a copy of the preprint by emailing the Biopolymers editorial office at biopolymers@wiley.com
Co-reporter:
Nature Structural and Molecular Biology 2002 9(3) pp:158 - 160
Publication Date(Web):
DOI:10.1038/nsb0302-158
Co-reporter:Amy Davidson, Darren W. Begley, Carmen Lau, Gabriele Varani
Journal of Molecular Biology (29 July 2011) Volume 410(Issue 5) pp:984-996
Publication Date(Web):29 July 2011
DOI:10.1016/j.jmb.2011.03.039
The HIV-1 transactivation response (TAR) element–Tat interaction is a potentially valuable target for treating HIV infection, but efforts to develop TAR-binding antiviral drugs have not yet yielded a successful candidate for clinical development. In this work, we describe a novel approach toward screening fragments against RNA that uses a chemical probe to target the Tat-binding region of TAR. This probe fulfills two critical roles in the screen: by locking the RNA into a conformation capable of binding other fragments, it simultaneously allows the identification of proximal binding fragments by ligand-based NMR. Using this approach, we have discovered six novel TAR-binding fragments, three of which were docked relative to the probe–RNA structure using experimental NMR restraints. The consistent orientations of functional groups in our data-driven docked structures and common electrostatic properties across all fragment leads reveal a surprising level of selectivity by our fragment-sized screening hits. These models further suggest linking strategies for the development of higher-affinity lead compounds for the inhibition of the TAR–Tat interaction.
Co-reporter:Mi-Kyung Lee, Angel Bottini, Meehyein Kim, Michael F. Bardaro, Ziming Zhang, Maurizio Pellecchia, Byong-Seok Choi and Gabriele Varani
Chemical Communications 2014 - vol. 50(Issue 3) pp:NaN370-370
Publication Date(Web):2013/10/29
DOI:10.1039/C3CC46973E
Through screening by NMR spectroscopy, we discovered a novel scaffold (DPQ: 6,7-dimethoxy-2-(1-piperazinyl)-4-quinazolinamine) that binds specifically to the influenza A virus RNA promoter. The solution structure of the RNA–DPQ complex reported here demonstrates that the internal loop is the binding site of DPQ. The scaffold exhibits antiviral activity against influenza viruses.
Co-reporter:Youhong Niu, Alisha “Jonesy” Jones, Haifan Wu, Gabriele Varani and Jianfeng Cai
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 19) pp:NaN6609-6609
Publication Date(Web):2011/07/01
DOI:10.1039/C1OB05738C
The interactions between proteins and RNAs are of vital importance for many cellular processes, including transcription and processing of RNA, translation, and viral infections. Here we report an γ-AApeptide that can mimic HIV-1 Tat protein and bind to TAR RNAs of HIV and BIV with nanomolar affinity, comparable to that of the RNA-binding fragment of Tat (amino acids 49–58). The interaction is resistant to the presence of a large excess of tRNA. With resistance to proteolytic hydrolysis and limitless potential for diversification, γ-AApeptides may emerge as a new class of peptidomimetics to modulate RNA-protein interactions.
Co-reporter:Mi-Kyung Lee, Angel Bottini, Meehyein Kim, Michael F. Bardaro, Ziming Zhang, Maurizio Pellecchia, Byong-Seok Choi and Gabriele Varani
Chemical Communications 2014 - vol. 50(Issue 83) pp:NaN12578-12578
Publication Date(Web):2014/09/12
DOI:10.1039/C4CC90361G
Correction for ‘A novel small-molecule binds to the influenza A virus RNA promoter and inhibits viral replication’ by Mi-Kyung Lee et al., Chem. Commun., 2014, 50, 368–370.
.jpg)









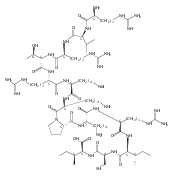
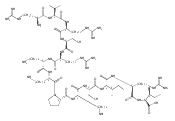
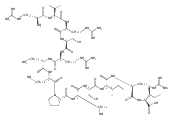
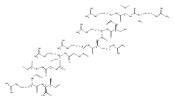











![N-(thien-2-ylmethyl)[1,2,4]triazolo[1,5-a]pyrimidine-2-carboxamide](http://img.cochemist.com/ccimg/510800/510712-33-1.png)
![N-(thien-2-ylmethyl)[1,2,4]triazolo[1,5-a]pyrimidine-2-carboxamide](http://img.cochemist.com/ccimg/510800/510712-33-1_b.png)





![tert-Butyl 4-[2-(aminomethyl)phenyl]piperazine-1-carboxylate](http://img.cochemist.com/ccimg/174900/174855-53-9.png)
![tert-Butyl 4-[2-(aminomethyl)phenyl]piperazine-1-carboxylate](http://img.cochemist.com/ccimg/174900/174855-53-9_b.png)


![Methyl 4-methylfuro[3,2-b]pyrrole-5-carboxylate](http://img.cochemist.com/ccimg/164700/164667-61-2.png)
![Methyl 4-methylfuro[3,2-b]pyrrole-5-carboxylate](http://img.cochemist.com/ccimg/164700/164667-61-2_b.png)











![Benzoic acid, 4-[([2,2'-bithiophen]-5-ylmethylene)amino]-](http://img.cochemist.com/ccimg/36000/35946-18-0.png)
![Benzoic acid, 4-[([2,2'-bithiophen]-5-ylmethylene)amino]-](http://img.cochemist.com/ccimg/36000/35946-18-0_b.png)
![[4-(2-Furyl)phenyl]methanol](http://img.cochemist.com/ccimg/18000/17920-85-3.png)
![[4-(2-Furyl)phenyl]methanol](http://img.cochemist.com/ccimg/18000/17920-85-3_b.png)


![[2,2'-Bithiophene]-5-methanol](http://img.cochemist.com/ccimg/3600/3515-30-8.png)
![[2,2'-Bithiophene]-5-methanol](http://img.cochemist.com/ccimg/3600/3515-30-8_b.png)