Co-reporter:Jing Guo, Mingyue Zhu, Tianxiao Wu, Chenzhou Hao, Kai Wang, Zizheng Yan, Wanxu Huang, Jian Wang, Dongmei Zhao, Maosheng Cheng
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 13(Issue 13) pp:
Publication Date(Web):1 July 2017
DOI:10.1016/j.bmc.2017.04.047
•Twenty-four indolin-2-one derivatives were synthesized.•All the compounds were screened for in vitro PAK4 inhibition assay.•Three of the compounds (11b, 12d, 12g) displayed promising enzymatic activity.•Compound 12g: Inducing apoptosis, G2/M phase arrest, Migration and Invasion inhibition by regulating PAK4-LIMK1-cofilin signalling pathway.•Docking and ADME profiling of 12g are reported.Utilizing a pharmacophore hybridization approach, a novel series of substituted indolin-2-one derivatives were designed, synthesized and evaluated for their in vitro biological activities against p21-activated kinase 4. Compounds 11b, 12d and 12g exhibited the most potent inhibitory activity against PAK4 (IC50 = 22 nM, 16 nM and 27 nM, respectively). Among them, compound 12g showed the highest antiproliferative activity against A549 cells (IC50 = 0.83 μM). Apoptosis analysis in A549 cells suggested that compound 12g delayed cell cycle progression by arresting cells in the G2/M phase of the cell cycle, retarding cell growth. Further investigation demonstrated that compound 12g strongly inhibited migration and invasion of A549 cells. Western blot analysis indicated that compound 12g potently inhibited the PAK4/LIMK1/cofilin signalling pathways. Finally, the binding mode between compound 12g with PAK4 was proposed by molecular docking. A preliminary ADME profile of the compound 12g was also drawn on the basis of QikProp predictions.Download high-res image (99KB)Download full-size image
Co-reporter:Chenzhou Hao, Xiaodong Li, Shuai Song, Bingyu Guo, Jing Guo, Jian Zhang, Qiaoling Zhang, Wanxu Huang, Jian Wang, Bin Lin, Maosheng Cheng, Feng Li and Dongmei Zhao
Organic & Biomolecular Chemistry 2016 vol. 14(Issue 32) pp:7676-7690
Publication Date(Web):15 Jul 2016
DOI:10.1039/C6OB01072E
A new series of novel 1-phenanthryl-tetrahydroisoquinoline derivatives were designed, synthesized and biologically evaluated for their PAK4 inhibitory activities and anti-proliferative effects against three cancer cell lines A549, MCF-7 and HT-1080. Among them, compound 12a exhibited the most potent inhibitory activity against PAK4 with an IC50 value of 0.42 μM. Moreover, this compound inhibited the invasion of A549 tumor cells by regulating the PAK4–LIMK1–cofilin signaling pathway in vitro, and exhibited anti-tumor activity in vivo in the A549 tumor xenograft model. To further evaluate the binding mode of 12a with PAK4, the biotinylated 12a derivative has been synthesized and it was used for immunoprecipitation assay. Intriguingly, our observations suggest that 12a interacts with both the N- and C-termini of PAK4.
Co-reporter:Shuai Song, Xiaodong Li, Jing Guo, Chenzhou Hao, Yan Feng, Bingyu Guo, Tongchao Liu, Qiaoling Zhang, Zhen Zhang, Ruijuan Li, Jian Wang, Bin Lin, Feng Li, Dongmei Zhao and Maosheng Cheng
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 12) pp:3803-3818
Publication Date(Web):09 Feb 2015
DOI:10.1039/C5OB00037H
Functional versatility and elevated expression in cancers have promoted p21-activated kinase 4 (PAK4) as one of the first-in-class anti-cancer drug targets. In this study, a series of novel 1-phenanthryl-tetrahydroisoquinoline analogues have been designed and synthesized as a novel class of small-molecule PAK4 inhibitors to fit into the cavity of PAK4. All of the target compounds were evaluated for their in vitro PAK4 inhibitory activities and antiproliferative activities. Lead optimization identified all the derivatives with more potency than the lead compound, especially compound 21a. Moreover, compound 21a significantly induced the cell cycle in the G1/S phase, and inhibited migration and invasion of MCF-7 cells via the regulation of the PAK4-LIMK1-cofilin signaling pathway. A molecular modeling study showed possible novel binding modes between 21a and PAK4 and provided a structural basis for further structure-guided design of PAK4 inhibitors.
Co-reporter:Rui-Juan Li;Jian Wang;Zhen Xu;Wan-Xu Huang;Jia Li;Sheng-Fei Jin; Dong-Mei Zhao; Mao-Sheng Cheng
ChemMedChem 2014 Volume 9( Issue 5) pp:1012-1022
Publication Date(Web):
DOI:10.1002/cmdc.201400016
Abstract
p21-Activated kinase 4 (PAK4) is a serine/threonine protein kinase that plays important roles in a wide variety of human diseases including cancer. Targeting this kinase with specific inhibitors is of great interest in the treatment of cancer. In this study, PAK4 and its interaction with ATP-competitive inhibitors was investigated by a combined ligand- and structure-based approach. First, a ligand-based pharmacophore model was generated, consisting of five chemical features: a positive ionizable center, two hydrophobic groups, a hydrogen bond donor, and a hydrogen bond acceptor, which is consistent with available SAR information. The characteristics of the active site were then described as a topological region and used in docking of nine selected inhibitors. Combination of the pharmacophore model and results from the docking studies allowed us to weigh the various pharmacophore features and to identify the positive ionizable center as a spacer rather than an essential point. This research led to the proposal of an interaction model inside the PAK4 active site and provided guidance for the design of more potent PAK4 inhibitors.
Co-reporter:Fengrong Li, Dongmei Zhao, Jinhong Ren, Feiyue Hao, Guyue Liu, Shengfei Jin, Yongkui Jing, Maosheng Cheng
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 11) pp:3256-3261
Publication Date(Web):1 June 2013
DOI:10.1016/j.bmc.2013.03.044
To develop new CYP26A1 inhibitors, a three-cycle virtual screening was carried out based on the constructed homology model of human CYP26A1 using Dock, Fred, Gold and AutoDock. Twenty-two compounds exhibited high scores and reasonable binding modes in molecular docking were purchased from Specs Company. Eighteen compounds were tested their abilities to enhance ATRA-induced differentiation in human acute promyelocytic leukemia NB4 cells. Eight of them enhanced the ability of ATRA to induce differentiation at concentrations of 0.5 and 1 μM. Among these compounds, 2-(2-methylfuran-3-carboxamido)-3-phenylpropanoic acid (S8) is of most effective in blocking ATRA breaking down in NB4 cells based on the LC–MS/MS assay.
Co-reporter:Chenzhou Hao, Wanxu Huang, Xiaodong Li, Jing Guo, Meng Chen, Zizheng Yan, Kai Wang, Xiaolin Jiang, Shuai Song, Jian Wang, Dongmei Zhao, Feng Li, Maosheng Cheng
European Journal of Medicinal Chemistry (5 May 2017) Volume 131() pp:
Publication Date(Web):5 May 2017
DOI:10.1016/j.ejmech.2017.02.063
•Using KY04031 as a starting compound, a library of 2, 4-diaminoquinazoline derivatives were designed and synthesized.•The compounds potently suppressed the enzymatic activities of PAK4 with IC50 values in the 10−6-10−8 M range.•The most potent compound, 9d, inhibited PAK4 kinase activity with an IC50 value of 0.033 μM.•Compound 9d inhibited the PAK4-driven cell proliferation in a dose-dependent manner.•Compound 9d potently suppressed cancer cell migration and invasion by regulating PAK4- LIMK1 signalling pathway.Upon analysis of the reported crystal structure of PAK4 inhibitor KY04031 (PAK4 IC50 = 0.790 μM) in the active site of PAK4, we investigated the possibility of changing the triazine core of KY04031 to a quinazoline. Using KY04031 as a starting compound, a library of 2, 4-diaminoquinazoline derivatives were designed and synthesized. These compounds were evaluated for PAK4 inhibition, leading to the identification of compound 9d (PAK4 IC50 = 0.033 μM). Compound 9d significantly induced the cell cycle in the G1/S phase and inhibited migration and invasion of A549 cells that over-express PAK4 via regulation of the PAK4-LIMK1 signalling pathway. A docking study of compound 9d was performed to elucidate its possible binding modes and to provide a structural basis for further structure-guided design of PAK4 inhibitors. Compound 9d may serve as a lead compound for anticancer drug discovery and as a valuable research probe for further biological investigation of PAK4.In this work, based on a cyclization strategy and structure-guided SAR design, a series of 2,4-diaminoquinazoline derivatives were developed as a new class of PAK4 inhibitors.
Co-reporter:Jian Zhang, Jian Wang, Qiqiang Guo, Yu Wang, Ying Zhou, Huizhi Peng, Maosheng Cheng, Dongmei Zhao, Feng Li
Cancer Letters (28 July 2014) Volume 349(Issue 2) pp:
Publication Date(Web):28 July 2014
DOI:10.1016/j.canlet.2013.09.028
Co-reporter:Chenzhou Hao, Xiaodong Li, Shuai Song, Bingyu Guo, Jing Guo, Jian Zhang, Qiaoling Zhang, Wanxu Huang, Jian Wang, Bin Lin, Maosheng Cheng, Feng Li and Dongmei Zhao
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 32) pp:NaN7690-7690
Publication Date(Web):2016/07/15
DOI:10.1039/C6OB01072E
A new series of novel 1-phenanthryl-tetrahydroisoquinoline derivatives were designed, synthesized and biologically evaluated for their PAK4 inhibitory activities and anti-proliferative effects against three cancer cell lines A549, MCF-7 and HT-1080. Among them, compound 12a exhibited the most potent inhibitory activity against PAK4 with an IC50 value of 0.42 μM. Moreover, this compound inhibited the invasion of A549 tumor cells by regulating the PAK4–LIMK1–cofilin signaling pathway in vitro, and exhibited anti-tumor activity in vivo in the A549 tumor xenograft model. To further evaluate the binding mode of 12a with PAK4, the biotinylated 12a derivative has been synthesized and it was used for immunoprecipitation assay. Intriguingly, our observations suggest that 12a interacts with both the N- and C-termini of PAK4.
Co-reporter:Shuai Song, Xiaodong Li, Jing Guo, Chenzhou Hao, Yan Feng, Bingyu Guo, Tongchao Liu, Qiaoling Zhang, Zhen Zhang, Ruijuan Li, Jian Wang, Bin Lin, Feng Li, Dongmei Zhao and Maosheng Cheng
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 12) pp:NaN3818-3818
Publication Date(Web):2015/02/09
DOI:10.1039/C5OB00037H
Functional versatility and elevated expression in cancers have promoted p21-activated kinase 4 (PAK4) as one of the first-in-class anti-cancer drug targets. In this study, a series of novel 1-phenanthryl-tetrahydroisoquinoline analogues have been designed and synthesized as a novel class of small-molecule PAK4 inhibitors to fit into the cavity of PAK4. All of the target compounds were evaluated for their in vitro PAK4 inhibitory activities and antiproliferative activities. Lead optimization identified all the derivatives with more potency than the lead compound, especially compound 21a. Moreover, compound 21a significantly induced the cell cycle in the G1/S phase, and inhibited migration and invasion of MCF-7 cells via the regulation of the PAK4-LIMK1-cofilin signaling pathway. A molecular modeling study showed possible novel binding modes between 21a and PAK4 and provided a structural basis for further structure-guided design of PAK4 inhibitors.

![1,3-Benzodioxole-5-acetic acid, α-[(3,4-dimethoxyphenyl)methylene]-, methyl ester, (αE)-](/data/chemimg/143100/1232031-45-6.png)
![1,3-Benzodioxole-5-acetic acid, α-[(3,4-dimethoxyphenyl)methylene]-, methyl ester, (αE)-](/data/chemimg/143100/1232031-45-6_b.png)
![(1S,4r)-4-(((S)-5-((3,5-bis(trifluoromethyl)benzyl)(2-methyl-2H-tetrazol-5-yl)amino)-7,9-dimethyl-2,3,4,5-tetrahydro-1H-benzo[b]azepin-1-yl)methyl)cyclohexanecarboxylic acid](http://img.cochemist.com/ccimg/1186500/1186486-62-3.png)
![(1S,4r)-4-(((S)-5-((3,5-bis(trifluoromethyl)benzyl)(2-methyl-2H-tetrazol-5-yl)amino)-7,9-dimethyl-2,3,4,5-tetrahydro-1H-benzo[b]azepin-1-yl)methyl)cyclohexanecarboxylic acid](http://img.cochemist.com/ccimg/1186500/1186486-62-3_b.png)
![2-[(5-tert-Butyl-2-methyl-furan-3-carbonyl)-amino]-3-phenyl-propionic acid](http://img.cochemist.com/ccimg/1397100/1397005-04-7.png)
![2-[(5-tert-Butyl-2-methyl-furan-3-carbonyl)-amino]-3-phenyl-propionic acid](http://img.cochemist.com/ccimg/1397100/1397005-04-7_b.png)
![1,3-Benzodioxole-5-acetic acid, α-[(3,4-dimethoxyphenyl)methylene]-, (αE)-](/data/chemimg/142900/1375207-25-2.png)
![1,3-Benzodioxole-5-acetic acid, α-[(3,4-dimethoxyphenyl)methylene]-, (αE)-](/data/chemimg/142900/1375207-25-2_b.png)


![Phenanthro[2,3-d][1,3]dioxole-6-carboxylic acid, 2,3-dimethoxy-](/data/chemimg/142900/1234172-49-6.png)
![Phenanthro[2,3-d][1,3]dioxole-6-carboxylic acid, 2,3-dimethoxy-](/data/chemimg/142900/1234172-49-6_b.png)
![Phenanthro[2,3-d][1,3]dioxole-5-carboxylic acid, 2,3-dimethoxy-](/data/chemimg/142900/1234172-48-5.png)
![Phenanthro[2,3-d][1,3]dioxole-5-carboxylic acid, 2,3-dimethoxy-](/data/chemimg/142900/1234172-48-5_b.png)
![Phenanthro[2,3-d][1,3]dioxole-6-carboxylic acid, 2,3-dimethoxy-, methyl ester](/data/chemimg/143100/1232031-51-4.png)
![Phenanthro[2,3-d][1,3]dioxole-6-carboxylic acid, 2,3-dimethoxy-, methyl ester](/data/chemimg/143100/1232031-51-4_b.png)
![6-Bromobenzo[d]thiazole-2-carboxylic acid](http://img.cochemist.com/ccimg/1188000/1187928-32-0.png)
![6-Bromobenzo[d]thiazole-2-carboxylic acid](http://img.cochemist.com/ccimg/1188000/1187928-32-0_b.png)
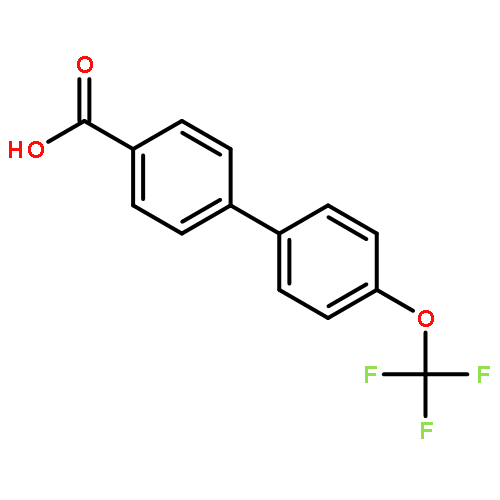
![2',5'-Difluoro-[1,1'-biphenyl]-4-carboxylic acid](http://img.cochemist.com/ccimg/920300/920294-24-2.png)
![2',5'-Difluoro-[1,1'-biphenyl]-4-carboxylic acid](http://img.cochemist.com/ccimg/920300/920294-24-2_b.png)
![(4S,5R)-5-[3,5-bis(trifluoromethyl)phenyl]-3-{[4'-fluoro-2'-methoxy-5'-(propan-2-yl)-4-(trifluoromethyl)biphenyl-2-yl]methyl}-4-methyl-1,3-oxazolidin-2-one](http://img.cochemist.com/ccimg/875500/875446-37-0.png)
![(4S,5R)-5-[3,5-bis(trifluoromethyl)phenyl]-3-{[4'-fluoro-2'-methoxy-5'-(propan-2-yl)-4-(trifluoromethyl)biphenyl-2-yl]methyl}-4-methyl-1,3-oxazolidin-2-one](http://img.cochemist.com/ccimg/875500/875446-37-0_b.png)



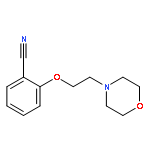
![[2-(2-Morpholinoethoxy)phenyl]methylamine](http://img.cochemist.com/ccimg/540800/540753-13-7.png)
![[2-(2-Morpholinoethoxy)phenyl]methylamine](http://img.cochemist.com/ccimg/540800/540753-13-7_b.png)
![3',4'-Difluoro-[1,1'-biphenyl]-4-carboxylic acid](http://img.cochemist.com/ccimg/505100/505082-81-5.png)
![3',4'-Difluoro-[1,1'-biphenyl]-4-carboxylic acid](http://img.cochemist.com/ccimg/505100/505082-81-5_b.png)
![6-Fluorobenzo[d]thiazole-2-carboxylic acid](http://img.cochemist.com/ccimg/479100/479028-67-6.png)
![6-Fluorobenzo[d]thiazole-2-carboxylic acid](http://img.cochemist.com/ccimg/479100/479028-67-6_b.png)



















![Propanethioic acid,2-methyl-, S-[2-[[[1-(2-ethylbutyl)cyclohexyl]carbonyl]amino]phenyl] ester](http://img.cochemist.com/ccimg/211600/211513-37-0.png)
![Propanethioic acid,2-methyl-, S-[2-[[[1-(2-ethylbutyl)cyclohexyl]carbonyl]amino]phenyl] ester](http://img.cochemist.com/ccimg/211600/211513-37-0_b.png)






![Benzeneacetic acid, α-[(3,4-dimethoxyphenyl)methylene]-3,4-dimethoxy-, (αZ)-](/data/chemimg/103200/172922-62-2.png)
![Benzeneacetic acid, α-[(3,4-dimethoxyphenyl)methylene]-3,4-dimethoxy-, (αZ)-](/data/chemimg/103200/172922-62-2_b.png)




![2-(3,5-DIMETHOXYPHENYL)-6-ETHYL-5-METHYLPYRAZOLO[1,5-A]PYRIMIDIN-7-OL](http://img.cochemist.com/ccimg/149800/149707-75-5.png)
![2-(3,5-DIMETHOXYPHENYL)-6-ETHYL-5-METHYLPYRAZOLO[1,5-A]PYRIMIDIN-7-OL](http://img.cochemist.com/ccimg/149800/149707-75-5_b.png)








![Methyl 6-chloro-3-oxo-3,4-dihydro-2H-benzo[b][1,4]oxazine-8-carboxylate](http://img.cochemist.com/ccimg/123100/123040-75-5.png)
![Methyl 6-chloro-3-oxo-3,4-dihydro-2H-benzo[b][1,4]oxazine-8-carboxylate](http://img.cochemist.com/ccimg/123100/123040-75-5_b.png)


![1,2-Benzenediamine, 4-[(3-chlorophenyl)-1H-imidazol-1-ylmethyl]-](http://img.cochemist.com/ccimg/122600/122558-90-1.png)
![1,2-Benzenediamine, 4-[(3-chlorophenyl)-1H-imidazol-1-ylmethyl]-](http://img.cochemist.com/ccimg/122600/122558-90-1_b.png)


![1H-Benzimidazole,6-[(3-chlorophenyl)-1H-imidazol-1-ylmethyl]-](http://img.cochemist.com/ccimg/115600/115575-11-6.png)
![1H-Benzimidazole,6-[(3-chlorophenyl)-1H-imidazol-1-ylmethyl]-](http://img.cochemist.com/ccimg/115600/115575-11-6_b.png)
![5-Methoxy-2-(4-methoxy-3,5-dimethylpyridin-2-yl)methylthio)-3H-imidazo[4,5-b]pyridine](http://img.cochemist.com/ccimg/113800/113713-24-9.png)
![5-Methoxy-2-(4-methoxy-3,5-dimethylpyridin-2-yl)methylthio)-3H-imidazo[4,5-b]pyridine](http://img.cochemist.com/ccimg/113800/113713-24-9_b.png)



![5,6,7,8-Tetrahydro-4-methoxy-6-methyl-1,3-dioxolo[4,5-g]isoquinolin-8-ol](http://img.cochemist.com/ccimg/108300/108261-07-0.png)
![5,6,7,8-Tetrahydro-4-methoxy-6-methyl-1,3-dioxolo[4,5-g]isoquinolin-8-ol](http://img.cochemist.com/ccimg/108300/108261-07-0_b.png)
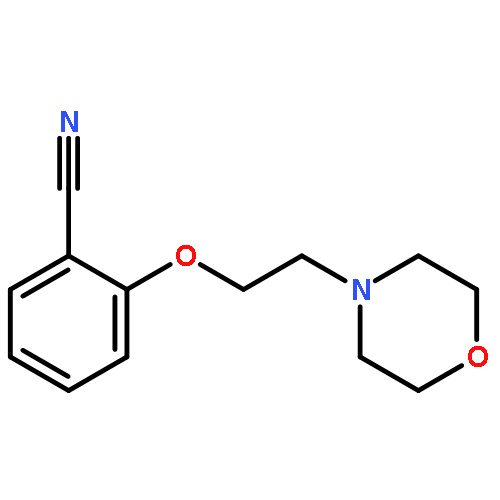
![Morpholine,4-[2-(2-nitrophenoxy)ethyl]-](http://img.cochemist.com/ccimg/105400/105337-21-1.png)
![Morpholine,4-[2-(2-nitrophenoxy)ethyl]-](http://img.cochemist.com/ccimg/105400/105337-21-1_b.png)


![2-Oxo-2-[[4-(trifluoromethyl)phenyl]amino]acetic AcidEthyl Ester](http://img.cochemist.com/ccimg/69100/69066-00-8.png)
![2-Oxo-2-[[4-(trifluoromethyl)phenyl]amino]acetic AcidEthyl Ester](http://img.cochemist.com/ccimg/69100/69066-00-8_b.png)








![Acetic acid, [(2-chlorophenyl)amino]oxo-, ethyl ester](http://img.cochemist.com/ccimg/53200/53117-16-1.png)
![Acetic acid, [(2-chlorophenyl)amino]oxo-, ethyl ester](http://img.cochemist.com/ccimg/53200/53117-16-1_b.png)
![Acetic acid, [(2-methylphenyl)amino]oxo-, ethyl ester](http://img.cochemist.com/ccimg/53200/53117-14-9.png)
![Acetic acid, [(2-methylphenyl)amino]oxo-, ethyl ester](http://img.cochemist.com/ccimg/53200/53117-14-9_b.png)














![1,3-Dioxolo[4,5-g]isoquinolinium,7,8-dihydro-4-methoxy-6-methyl-, iodide (1:1)](http://img.cochemist.com/ccimg/31000/30936-27-7.png)
![1,3-Dioxolo[4,5-g]isoquinolinium,7,8-dihydro-4-methoxy-6-methyl-, iodide (1:1)](http://img.cochemist.com/ccimg/31000/30936-27-7_b.png)







![Acetic acid, [(4-bromophenyl)amino]oxo-, ethyl ester](http://img.cochemist.com/ccimg/24500/24451-15-8.png)
![Acetic acid, [(4-bromophenyl)amino]oxo-, ethyl ester](http://img.cochemist.com/ccimg/24500/24451-15-8_b.png)




![Acetic acid,2-[(4-methylphenyl)amino]-2-oxo-, ethyl ester](http://img.cochemist.com/ccimg/18600/18522-98-0.png)
![Acetic acid,2-[(4-methylphenyl)amino]-2-oxo-, ethyl ester](http://img.cochemist.com/ccimg/18600/18522-98-0_b.png)


![(3R)-6,7-dimethoxy-3-[(5S)-4-methoxy-6-methyl-5,6,7,8-tetrahydro[1,3]dioxolo[4,5-g]isoquinolin-5-yl]-2-benzofuran-1(3H)-one](http://img.cochemist.com/ccimg/6100/6035-40-1.png)
![(3R)-6,7-dimethoxy-3-[(5S)-4-methoxy-6-methyl-5,6,7,8-tetrahydro[1,3]dioxolo[4,5-g]isoquinolin-5-yl]-2-benzofuran-1(3H)-one](http://img.cochemist.com/ccimg/6100/6035-40-1_b.png)
![4'-Fluoro-[1,1'-biphenyl]-4-carboxylic acid](http://img.cochemist.com/ccimg/5800/5731-10-2.png)
![4'-Fluoro-[1,1'-biphenyl]-4-carboxylic acid](http://img.cochemist.com/ccimg/5800/5731-10-2_b.png)


![Acetic acid,2-[(4-chlorophenyl)amino]-2-oxo-, ethyl ester](http://img.cochemist.com/ccimg/5400/5397-14-8.png)
![Acetic acid,2-[(4-chlorophenyl)amino]-2-oxo-, ethyl ester](http://img.cochemist.com/ccimg/5400/5397-14-8_b.png)
![4-(4,4,5,5-TETRAMETHYL-1,3,2-DIOXABOROLAN-2-YL)-2-{[4-(TRIFLUOROM<WBR />ETHYL)BENZYL]OXY}PYRIDINE](http://img.cochemist.com/ccimg/3700/3622-03-5.png)
![4-(4,4,5,5-TETRAMETHYL-1,3,2-DIOXABOROLAN-2-YL)-2-{[4-(TRIFLUOROM<WBR />ETHYL)BENZYL]OXY}PYRIDINE](http://img.cochemist.com/ccimg/3700/3622-03-5_b.png)









![6-Methoxybenzo[d]thiazole-2-carboxylic acid](http://img.cochemist.com/ccimg/1000/946-13-4.png)
![6-Methoxybenzo[d]thiazole-2-carboxylic acid](http://img.cochemist.com/ccimg/1000/946-13-4_b.png)

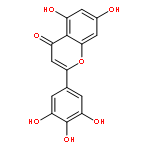

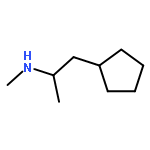
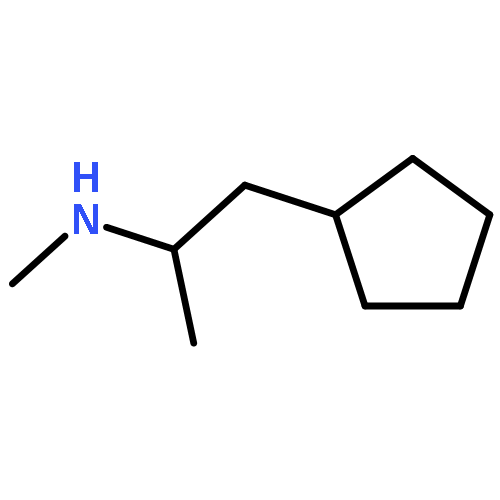
![1,3-Dioxolo[4,5-g]isoquinolin-5-ol,5,6,7,8-tetrahydro-4-methoxy-6-methyl-](http://img.cochemist.com/ccimg/100/82-54-2.png)
![1,3-Dioxolo[4,5-g]isoquinolin-5-ol,5,6,7,8-tetrahydro-4-methoxy-6-methyl-](http://img.cochemist.com/ccimg/100/82-54-2_b.png)


![1H-PYRROLO[3,2-B]PYRIDINE-2-CARBOXAMIDE](http://img.cochemist.com/ccimg/317900/317806-87-4.png)
![1H-PYRROLO[3,2-B]PYRIDINE-2-CARBOXAMIDE](http://img.cochemist.com/ccimg/317900/317806-87-4_b.png)
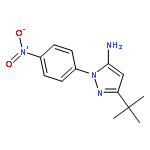
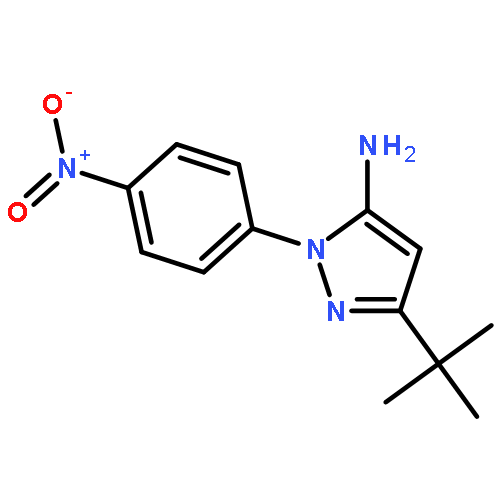


![3-nitro-4-(phenylmethoxy)phenyl]-oxirane](http://img.cochemist.com/ccimg/51600/51582-41-3.png)
![3-nitro-4-(phenylmethoxy)phenyl]-oxirane](http://img.cochemist.com/ccimg/51600/51582-41-3_b.png)
![[(2s)-2-(3-formamido-4-hydroxyphenyl)-2-hydroxyethyl]-[(2s)-1-(4-methoxyphenyl)propan-2-yl]azanium](http://img.cochemist.com/ccimg/43300/43229-80-7.png)
![[(2s)-2-(3-formamido-4-hydroxyphenyl)-2-hydroxyethyl]-[(2s)-1-(4-methoxyphenyl)propan-2-yl]azanium](http://img.cochemist.com/ccimg/43300/43229-80-7_b.png)