Co-reporter:Ryuji Uchida, Daiki Lee, Ibuki Suwa, Masaki Ohtawa, Nozomu Watanabe, Ayumu Demachi, Satoshi Ohte, Takenobu Katagiri, Tohru Nagamitsu, and Hiroshi Tomoda
Organic Letters November 3, 2017 Volume 19(Issue 21) pp:5980-5980
Publication Date(Web):October 24, 2017
DOI:10.1021/acs.orglett.7b03003
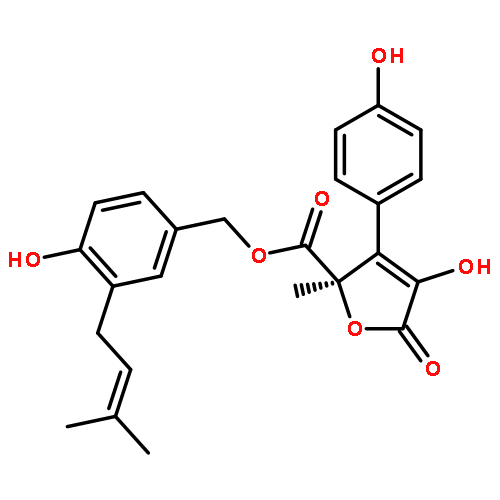
Three new compounds, designated scopranones A–C, were isolated from the culture broth of a soil isolate, Streptomyces sp. BYK-11038, and shown to be inhibitors of bone morphogenetic protein (BMP) induced alkaline phosphatase activity in a BMP receptor mutant cell line. The structures were elucidated using NMR and other spectral data. The scopranones have an unusual structure with two atypical scooplike moieties linked at the tails to form part of a unique 3-furanone ring.
Co-reporter:Hiroyuki Ishijima, Ryuji Uchida, Masaki Ohtawa, Ariko Kondo, Kenichiro Nagai, Keisuke Shima, Kenichi Nonaka, Rokuro Masuma, Susumu Iwamoto, Hideyuki Onodera, Tohru Nagamitsu, and Hiroshi Tomoda
The Journal of Organic Chemistry 2016 Volume 81(Issue 17) pp:7373-7383
Publication Date(Web):July 11, 2016
DOI:10.1021/acs.joc.6b00952
The targets of antifungal antibiotics in clinical use are more limited than those of antibacterial antibiotics. Therefore, new antifungal antibiotics with different mechanisms of action are desired. In the course of our screening for antifungal antibiotics of microbial origins, new antifungal antibiotics, simplifungin (1) and valsafungins A (2) and B (3), were isolated from cultures of the fungal strains Simplicillium minatense FKI-4981 and Valsaceae sp. FKH-53, respectively. The structures of 1 to 3 including their absolute stereochemistries were elucidated using various spectral analyses including NMR and collision-induced dissociation (CID)-MS/MS as well as chemical approaches including modifications to the Mosher’s method. They were structurally related to myriocin. They inhibited the growth of yeast-like and zygomycetous fungi with MICs ranging between 0.125 and 8.0 μg/mL. An examination of their mechanisms of action by the newly established assay using LC–MS revealed that 1 and 2 inhibited serine palmitoyltransferase activity, which is involved in sphingolipid biosynthesis, with IC50 values of 224 and 24 nM, respectively.
Co-reporter:Junji Inokoshi, Yuichiro Nakamura, Saori Komada, Katsuichiro Komatsu, Hideaki Umeyama and Hiroshi Tomoda
The Journal of Antibiotics 2016 69(11) pp:798-805
Publication Date(Web):April 6, 2016
DOI:10.1038/ja.2016.35
Viridicatumtoxin and spirohexaline, small fungal molecules with a tetracyclic scaffold and an additional spirobicyclic ring in common, were found to inhibit bacterial undecaprenyl pyrophosphate (UPP) synthase with IC50 values of 4 and 9 μm, respectively. These molecules showed weak inhibitory activity against catalytically related enzymes such as bacterial octaprenyl pyrophosphate synthase and yeast dehydrodolichyl pyrophosphate synthase, indicating that the compounds preferentially inhibit UPP synthase. They showed antimicrobial activity, particularly against Gram-positive bacteria including methicillin-resistant Staphylococcus aureus (MRSA). Furthermore, molecular modeling strongly suggested that the hydrophobic spirobicyclic ring of viridicatumtoxin interacts with three hydrophobic clefts of the active site in MRSA UPP synthase.
Co-reporter:Ryuji Uchida, Kento Nakajyo, Keisuke Kobayashi, Taichi Ohshiro, Takeshi Terahara, Chiaki Imada and Hiroshi Tomoda
The Journal of Antibiotics 2016 69(8) pp:647-651
Publication Date(Web):March 16, 2016
DOI:10.1038/ja.2016.27
A new depsidone, named 7-chlorofolipastatin, and five known structurally related depsidones were isolated from the culture broth of the marine-derived fungus Aspergillus ungui NKM-007 by solvent extraction and HPLC using an octadecylsilyl column. The structure of 7-chlorofolipastatin was elucidated by various spectroscopic data including 1D and 2D NMR spectroscopy. 7-Chlorofolipastatin inhibited sterol O-acyltransferase (SOAT) 1 and 2 isozymes in cell-based and enzyme assays using SOAT1- and SOAT2-expressing Chinese hamster ovary (CHO) cells.
Co-reporter:Daisuke Yamamuro, Ryuji Uchida, Masaki Ohtawa, Shiho Arima, Yushi Futamura, Masumi Katane, Hiroshi Homma, Tohru Nagamitsu, Hiroyuki Osada, Hiroshi Tomoda
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 2) pp:313-316
Publication Date(Web):15 January 2015
DOI:10.1016/j.bmcl.2014.11.042
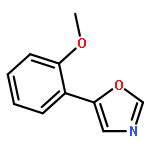
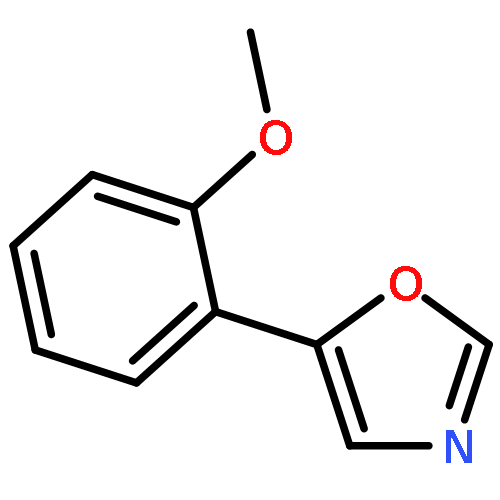
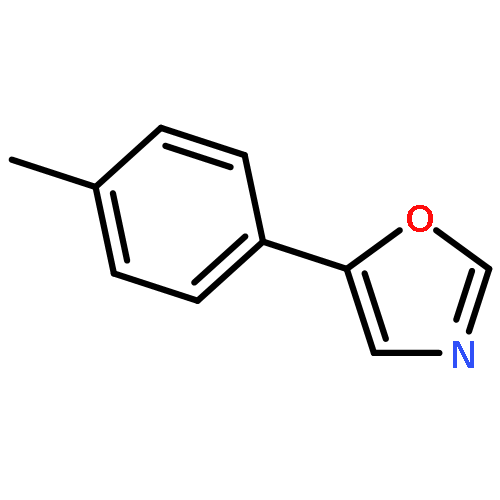
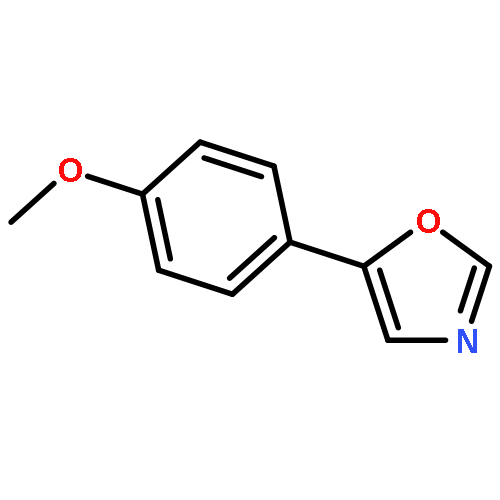
5-(4′-Methoxyphenyl)-oxazole (MPO), originally reported as a synthetic compound, was isolated from fungal culture broth as an inhibitor of hatch and growth of Caenorhabditis elegans. Nineteen MPO derivatives were chemically synthesized, but showed no effect on C. elegans hatch and growth. These findings strongly suggested that the whole structure of MPO is essential for anti-C. elegans activity.
Co-reporter:Keisuke Kobayashi, Nobuaki Tsukasaki, Ryuji Uchida, Yuichi Yamaguchi and Hiroshi Tomoda
The Journal of Antibiotics 2015 68(10) pp:615-619
Publication Date(Web):April 22, 2015
DOI:10.1038/ja.2015.37
A new compound designated as clonoamide was isolated from a culture broth of the fungus Clonostachys sp. BF-0131 by solvent extraction, Diaion HP20 column chromatography, octadecylsilyl column chromatography and preparative HPLC as an inhibitor of sterol O-acyltransferase (SOAT). The structure of clonoamide was elucidated as 2-oxo-9E,11E-tridecandienyl acetamide by various spectral analyses including NMR. The compound inhibited SOAT1 and SOAT2 isozymes with IC50 values of 39 and 110 μm, respectively, in a cell-based assay using SOAT1- and SOAT2-expressing Chinese hamster ovary cells.
Co-reporter:Takashi Fukuda, Minori Shinkai, Eri Sasaki, Kenichiro Nagai, Yuko Kurihara, Akihiko Kanamoto and Hiroshi Tomoda
The Journal of Antibiotics 2015 68(10) pp:620-627
Publication Date(Web):April 22, 2015
DOI:10.1038/ja.2015.41
Eight new thiodiketopiperazines, designated as graphiumins A to H (1–8), were isolated along with bisdethiobis(methylthio)-deacetylaranotin (9) and bisdethiobis(methylthio)-deacetylapoaranotin (10) from the culture broth of the marine-derived fungus Graphium sp. OPMF00224. The structures of the graphiumins were elucidated based on spectroscopic analyses (1D and 2D NMR data, ROESY correlations and CD data) and chemical methods. The absolute configuration of the common (3S)-3-hydroxy-octanoyl acid residue in 1, 3 and 4 was determined by hydrolysis, benzoyl derivatization and HPLC analysis using a chiral column. Five graphiumins moderately inhibited yellow pigment production by methicillin-resistant Staphylococcus aureus.
Co-reporter:Keisuke Kobayashi, Takashi Fukuda, Takeo Usui, Yuko Kurihara, Akihiko Kanamoto and Hiroshi Tomoda
The Journal of Antibiotics 2015 68(2) pp:126-132
Publication Date(Web):August 6, 2014
DOI:10.1038/ja.2014.100
Marine-derived Streptomyces sp. OPMA00072 was found to produce inhibitors of the synthesis of neutral lipids in a cell-based assay using Chinese hamster ovary (CHO) cells. A new 16-membered macrolide named bafilomycin L (BFL) (1) was isolated along with the known structurally related bafilomycin C1 (BFC1) (3) from the culture broth of the actinomycete by solvent extraction, octadecylsilyl column chromatography and HPLC. BFL inhibited cholesteryl ester (CE) synthesis in CHO cells with an IC50 value of 0.83 nM and also in mouse peritoneal macrophages with an IC50 of 6.1 nM. In addition, BFL blocked cellular acidification in HeLa cells by interfering with vacuolar H+-ATPase (V-ATPase) as well as other bafilomycins. These data strongly suggest that BFL disturbed the lysosome function to block cholesterol metabolism, leading to the inhibition of CE accumulation in mammalian cells.
Co-reporter:Daisuke Matsuda, Taichi Ohshiro, Masaki Ohtawa, Hiroyuki Yamazaki, Tohru Nagamitsu and Hiroshi Tomoda
The Journal of Antibiotics 2015 68(1) pp:27-34
Publication Date(Web):July 9, 2014
DOI:10.1038/ja.2014.91
Pyripyropene A (PPPA, 1) of fungal origin, a selective inhibitor of acyl-CoA:cholesterol acyltransferase 2 (ACAT2), proved orally active in atherogenic mouse models. The in vitro metabolites of 1 in liver microsomes and plasma of human, rabbit, rat and mouse were analyzed by ultra fast liquid chromatography and liquid chromatography/tandem mass spectrometry. In the liver microsomes from all species, successive hydrolysis occurred at the 1-O-acetyl residue, then at the 11-O-acetyl residue of 1, while the 7-O-acetyl residue was resistant to hydrolysis. Furthermore, dehydrogenation of the newly generated 11-alcoholic hydroxyl residue occurred in human and mouse-liver microsomes, while oxidation of the pyridine ring occurred in human and rabbit liver microsomes. On the other hand, hydrolysis of the 7-O-acetyl residue proceeded only in the mouse plasma. These data indicated that the in vitro metabolic profiles of 1 have subtle differences among animal species. All of the PPPA metabolites observed in liver microsomes and plasma markedly decreased ACAT2 inhibitory activity. These findings will help us to synthesize new PPPA derivatives more effective in in vivo study than 1.
Co-reporter:Ryuji Uchida, Sayaka Yokota and Hiroshi Tomoda
The Journal of Antibiotics 2014 67(11) pp:783-786
Publication Date(Web):June 11, 2014
DOI:10.1038/ja.2014.71
A novel abrogator of bleomycin-induced G2 arrest in Jurkat cells, habiterpenol (1), was isolated from the culture broth of Phytohabitans suffuscus 3787_5. The planar structure of 1 was elucidated by spectroscopic study (1D and 2D NMR, MS, UV and IR), and the relative stereochemistry was elucidated by ROESY experiments. Compound 1 belongs to a pentacyclic meroterpenoid having a labdan-type diterpene connecting to an indane moiety.
Co-reporter:Takashi Fukuda, Kenta Shimoyama, Tohru Nagamitsu and Hiroshi Tomoda
The Journal of Antibiotics 2014 67(6) pp:445-450
Publication Date(Web):March 19, 2014
DOI:10.1038/ja.2014.14
Citridone A (1), originally isolated as a potentiator of antifungal miconazole activity from a fungal culture broth, has a phenyl-R-furopyridone structure. Because of its unique ring structure, 11 derivatives were chemically synthesized and their biological activity was evaluated. Derivatives 17, 20 and 21 potentiated miconazole activity against Candida albicans. Furthermore, 1, 14, 20 and 21 were found to inhibit yellow pigment production in methicillin-resistant Staphylococcus aureus.
Co-reporter:Ryuji Uchida, Sayaka Yokota, Daisuke Matsuda, Atsuko Matsumoto, Susumu Iwamoto, Hideyuki Onodera, Yoko Takahashi and Hiroshi Tomoda
The Journal of Antibiotics 2014 67(11) pp:777-781
Publication Date(Web):June 11, 2014
DOI:10.1038/ja.2014.62
A small molecule named habiterpenol produced by actinomycete Phytohabitans suffuscus 3787_5 was found to abrogate bleomycin-induced G2 arrest in Jurkat cells. Habiterpenol showed no cytotoxic effect on Jurkat cells even at 273 μM; however, the compound inhibited bleomycin-induced G2 arrest in Jurkat cells with an IC50 value of 3.55 μM, while it showed no effect on colchicine-induced M arrest even at 273 μM. These results indicated that habiterpenol selectively abrogated bleomycin-induced G2 arrest in Jurkat cells.
Co-reporter:Masaki Ohtawa, Hiroyuki Yamazaki, Daisuke Matsuda, Taichi Ohshiro, Lawrence L. Rudel, Satoshi Ōmura, Hiroshi Tomoda, Tohru Nagamitsu
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 9) pp:2659-2662
Publication Date(Web):1 May 2013
DOI:10.1016/j.bmcl.2013.02.088
Synthesis and structure–activity relationships of 7-O-p-cyanobenzoyl pyripyropene A derivatives with modification at C1 and 11 are described. Regioselective mono-deprotection of di-tert-butylsilylene acetal was critical in their synthesis.
Co-reporter:Nobuhiro Koyama, Yuriko Tokura, Yoko Takahashi, Hiroshi Tomoda
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 3) pp:860-863
Publication Date(Web):1 February 2013
DOI:10.1016/j.bmcl.2012.11.044
A new actinomycete metabolite designated nosokophic acid was isolated from the culture broth of nosokomycin-producing Streptomyces sp. K04-0144, and the structure was elucidated by various NMR experiments. Nosokophic acid was found to be 3-phosphoglycosyl-2-sesquiterpenyl dihydroxypropionic acid, a predicted biosynthetic intermediate of nosokomycin-related moenomycins. The compound showed no activity against MRSA, but potentiated imipenem activity against MRSA by 512-fold.
Co-reporter:Masaki Ohtawa, Hiroyuki Yamazaki, Satoshi Ohte, Daisuke Matsuda, Taichi Ohshiro, Lawrence L. Rudel, Satoshi Ōmura, Hiroshi Tomoda, Tohru Nagamitsu
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 5) pp:1285-1287
Publication Date(Web):1 March 2013
DOI:10.1016/j.bmcl.2012.12.099
In an effort to develop potent and selective inhibitors toward ACAT2, structure–activity relationship studies were carried out using derivatives based on pyripyropene A (PPPA, 1). We have successfully developed novel PPPA derivatives with a 7-O-substituted benzoyl substituent that significantly exhibit more potent ACAT2 inhibitory activity and higher ACAT2 isozyme selectivity than 1.
Co-reporter:Masaki Ohtawa, Hiroyuki Yamazaki, Satoshi Ohte, Daisuke Matsuda, Taichi Ohshiro, Lawrence L. Rudel, Satoshi Ōmura, Hiroshi Tomoda, Tohru Nagamitsu
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 13) pp:3798-3801
Publication Date(Web):1 July 2013
DOI:10.1016/j.bmcl.2013.04.075
In an effort to develop potent and selective inhibitors toward ACAT2, structure–activity relationship studies were carried out using derivatives based on pyripyropene A (PPPA, 1). In particular, we investigated the possibility of introducing appropriate 1,11-O-benzylidene and 7-O-substituted benzoyl moieties into PPPA (1). The new o-substituted benzylidene derivatives showed higher selectivity for ACAT2 than PPPA (1). Among them, 1,11-O-o-methylbenzylidene-7-O-p-cyanobenzoyl PPPA derivative 7q and 1,11-O-o,o-dimethylbenzylidene-7-O-p-cyanobenzoyl PPPA derivative 7z proved to be potent ACAT2 inhibitors with unprecedented high isozyme selectivity.
Co-reporter:Junji Inokoshi, Naoki Shigeta, Takashi Fukuda, Ryuji Uchida, Kenichi Nonaka, Rokurou Masuma and Hiroshi Tomoda
The Journal of Antibiotics 2013 66(9) pp:549-554
Publication Date(Web):May 29, 2013
DOI:10.1038/ja.2013.44
A new compound, designated epi-trichosetin (1), was isolated along with the known compound trichosetin (2) from the culture broth of Fusarium oxysporum FKI-4553 by solvent extraction, silica gel column chromatography and reversed-phase HPLC. The structure of 1 was elucidated by comparing various spectral data with those of 2, revealing that 1 was a stereoisomer of 2. Compounds 1 and 2 inhibited the undecaprenyl pyrophosphate synthase activity of Staphylococcus aureus with IC50 values of 83 and 30 μM, respectively, and showed antimicrobial activity, particularly against Gram-positive bacteria, including methicillin-sensitive and -resistant S. aureus.
Co-reporter:Mio Kawaguchi, Ryuji Uchida, Satoshi Ohte, Natsuki Miyachi, Keisuke Kobayashi, Noriko Sato, Kenichi Nonaka, Rokuro Masuma, Takashi Fukuda, Tadashi Yasuhara and Hiroshi Tomoda
The Journal of Antibiotics 2013 66(3) pp:179-189
Publication Date(Web):2013-03-01
DOI:10.1038/ja.2012.127
Eight new dinapinones, AB1, AB2, AC1, AC2, AD1, AD2, AE1 and AE2, were isolated from the culture broth of Talaromyces pinophilus FKI-3864. The structures of these dinapinones were elucidated by various NMR experiments. All these dinapinones possessed the same biaryl dihydronaphthopyranone skeleton consisting of a heterodimer with one monapinone A and one different monapinone. Dinapinones AB1 and AB2, consisting of monapinones A and B, were atropisomers. Similarly, dinapinones AC1 and AC2, consisting of monapinones A and C, dinapinones AD1 and AD2, consisting of monapinones A and D, and dinapinones AE1 and AE2, consisting of monapinones A and E, were atropisomers. Dinapinone AB2 showed potent inhibition of triacylglycerol (TG) synthesis in intact mammalian cells with an IC50 value of 1.17 μM, whereas the other dinapinones showed weak inhibition of TG synthesis.
Co-reporter:Mio Kawaguchi, Kenichi Nonaka, Rokuro Masuma and Hiroshi Tomoda
The Journal of Antibiotics 2013 66(1) pp:17-21
Publication Date(Web):November 14, 2012
DOI:10.1038/ja.2012.79
A convenient and efficient method was established for isolating antifungal antibiotic-producing fungi from soil samples. In this method, soil samples were diluted and directly plated in agar medium by the standard fungi-isolating method, and the plates were cultured at 27 °C for 2–3 days to permit the growth of fungal colonies. Then, the suspension of pathogenic Candida albicans in saline (40 μl, 5–10 × 105 CFU ml−1) was overlaid by spraying on the plates under controlled conditions in the safety cabinet. After 1-day incubation, fungal colonies showing an antagonistic effect with the inhibition zone against sprayed C. albicans were selected. Among 151 isolates, 26 strains were found to reproduce anti-C. albicans activity in liquid medium, yielding a higher selection rate (17.2%) than that (3.1%) by the traditional method. This new method can be applied for isolation of microorganisms (fungi and actinomycetes) that produce antibiotics active against pathogenic microorganisms.
Co-reporter:Mio Kawaguchi, Takashi Fukuda, Ryuji Uchida, Kenichi Nonaka, Rokuro Masuma and Hiroshi Tomoda
The Journal of Antibiotics 2013 66(1) pp:23-29
Publication Date(Web):November 21, 2012
DOI:10.1038/ja.2012.75
Cylindrol A5, a new ascochlorin congener, was isolated along with 14 known compounds from the culture broth of Cylindrocarpon sp. FKI-4602 by solvent extraction, octadecylsilane column chromatography and HPLC. The structure of cylindrol A5 was elucidated by spectral analyses, including NMR. The compound has an ascochlorin skeleton consisting of a resorcin aldehyde and a cyclohexanone moieties. Cylindrol A5 showed moderate antimicrobial activity against Bacillus subtilis, Kocuria rhizophila, Mycobacterium smegmatis and Acholeplasma laidlawii. The biosynthetic pathway to cylindrol A5 was deduced from the 14 isolated metabolites of the fungal strain.
Co-reporter:Junji Inokoshi, Yuichiro Nakamura, Zhang Hongbin, Ryuji Uchida, Ken-ichi Nonaka, Rokuro Masuma and Hiroshi Tomoda
The Journal of Antibiotics 2013 66(1) pp:37-41
Publication Date(Web):November 21, 2012
DOI:10.1038/ja.2012.83
An enzyme assay for bacterial undecaprenyl pyrophosphate (UPP) synthase was performed to screen microbial culture broths for inhibitors of UPP synthase. During the course of this screening program, an EtOH extract of a rice culture of Penicillium brasilianum FKI-3368 was found to inhibit UPP synthase activity. From activity-guided purification, a new compound-designated spirohexaline was isolated together with the structurally related and known viridicatumtoxin by ethyl acetate extraction silica gel and octadecylsilane column chromatographies and high-performance liquid chromatography. The structure of spirohexaline was elucidated by spectroscopic analysis, including NMR. Spirohexaline and viridicatumtoxin have a common hexacycline structure produced by fusion of a tetracycline-type ring with a spiro-type ring. They inhibited UPP synthase activity with IC50 values of 9.0 and 4.0 μM, respectively.
Co-reporter:Taichi Ohshiro, Daisuke Matsuda, Takeuchi Kazuhiro, Ryuji Uchida, Kenichi Nonaka, Rokuro Masuma and Hiroshi Tomoda
The Journal of Antibiotics 2012 65(5) pp:255-262
Publication Date(Web):March 14, 2012
DOI:10.1038/ja.2012.12
Verticillium sp. FKI-2679, a soil isolate, was found to produce inhibitors of acyl-CoA:cholesterol acyltransferase (ACAT) in a cell-based assay using ACAT1- and ACAT2-expressing CHO cells. Three new compounds, verticilides A2, A3 and B1, were isolated along with a known compound, verticilide A1, from the fermentation broth of the fungus by solvent extraction, ODS column chromatography, silica gel column chromatography and preparative HPLC. Structure elucidation showed that these compounds were new cyclic depsipeptide. Verticilides A1, A2, A3 and B1 showed a degree of selectivity towards ACAT2, with IC50s 8.5–11-fold more potent than observed against ACAT1.
Co-reporter:Narihiro Ugaki, Daisuke Matsuda, Hiroyuki Yamazaki, Kenichi Nonaka, Rokuro Masuma, Satoshi Ōmura and Hiroshi Tomoda
The Journal of Antibiotics 2012 65(1) pp:15-19
Publication Date(Web):November 23, 2011
DOI:10.1038/ja.2011.105
A new bis-naphtho-γ-pyrone isomer named isochaetochromin A1 was isolated along with known isochaetochromins B1 and B2 from the culture broth of Penicillium sp. FKI-4942 by solvent extraction, silica gel column chromatography and HPLC. Among them, isochaetochromin B1 showed the most potent inhibitory activity of triacylglycerol synthesis with an IC50 value of 5.6 μM, followed by isochaetochromins B2 (IC50, 11 μM) and A1 (33 μM).
Co-reporter:Narihiro Ugaki, Hiroyuki Yamazaki, Ryuji Uchida and Hiroshi Tomoda
The Journal of Antibiotics 2012 65(1) pp:21-24
Publication Date(Web):November 30, 2011
DOI:10.1038/ja.2011.106
The structure of a new congener of chaetochromin, an inhibitor of triacylglycerol synthesis in CHO-K1 cells produced by Penicillium sp. FKI-4942, was elucidated by spectroscopic methods, including various NMR experiments. Isochaetochromin A1 has a bis-naphtho-γ-pyrone moiety.
Co-reporter:Takashi Fukuda, Ryuji Uchida, Satoshi Ohte, Hiroyo Inoue, Hiroyuki Yamazaki, Daisuke Matsuda, Kenichi Nonaka, Rokurou Masuma, Takenobu Katagiri and Hiroshi Tomoda
The Journal of Antibiotics 2012 65(11) pp:565-569
Publication Date(Web):September 5, 2012
DOI:10.1038/ja.2012.70
Two new butenolides, designated trichocyalides A and B, were isolated along with the known compound harzianolide, from the culture broth of Trichoderma sp. FKI-5513 by solvent extraction, ODS column chromatography and HPLC. Their structures were elucidated by several spectral analyses, showing that they have the common skeleton of butenofuranone. Trichocyalides A and B inhibited alkaline phosphatase (ALP) activity, a typical marker enzyme of osteoblastic differentiation (IC50: 83.0 and 187 μM, respectively), in bone morphogenetic protein (BMP)-stimulated C2C12 myoblasts mutant cells, which stably express BMP receptor activity, whereas harzianolide showed no inhibitory activity against ALP even at 500 μM.
Co-reporter:Ryuji Uchida, Satoshi Ohte, Kyosuke Kawamoto, Hiroyuki Yamazaki, Mio Kawaguchi and Hiroshi Tomoda
The Journal of Antibiotics 2012 65(8) pp:419-425
Publication Date(Web):May 30, 2012
DOI:10.1038/ja.2012.41
During our screening program for microbial inhibitors of triacylglycerol synthesis in mammalian cells, four structurally related new compounds, dinapinones A1 (1) and A2 (2) and monapinones A (3) and B (4), were isolated from the culture broth of Penicillium pinophilum FKI-3864. Compounds 3 and 4 were produced by the fungus only when fermented in seawater-supplemented medium. The structures of 1 to 4 were elucidated by spectroscopic studies including various NMR experiments. Compounds 1 and 2 were atropisomers consisting of two monomers with the same planar structure of dihydronaphthopyranone as 3. The absolute stereochemistry of 3 was elucidated by NOE experiment and circular dichroism spectra. Furthermore, the stereochemistry of 1 and 2 was elucidated by in vitro conversion from the structure-defined 3 to its dimers 1 and 2.
Co-reporter:Junji Inokoshi;Maki Matsuhama;Midori Miyake
Applied Microbiology and Biotechnology 2012 Volume 95( Issue 2) pp:451-460
Publication Date(Web):2012 July
DOI:10.1007/s00253-012-3973-8
The biosynthetic gene cluster for lariatins A and B, anti-mycobacterial peptide antibiotics with a unique “lasso” structure, was cloned from Gram-positive bacterium Rhodococcus jostii K01-B0171. Random transposition mutagenesis using IS1415 derivative was carried out to identify a chromosomal locus involved in lariatin biosynthesis and six independent lariatin non-producing variants were obtained. Arbitrary PCR revealed that one insertion was located near the region involved in lariatin biosynthesis. Using the lariatin gene as a probe, a genomic library of R. jostii K01-B0171 was screened by colony hybridization, and two clones were obtained. Sequence analysis of these clones revealed that the gene cluster for lariatin biosynthesis spanning about 4.5 kb consisted of five open reading frames (larA to larE). We proposed that the linear precursor LarA is processed by LarB, LarC, and LarD, and the mature lariatin is exported by LarE.
Co-reporter:Satoshi Ohte, Daisuke Matsuda, Ryuji Uchida, Kenichi Nonaka, Rokuro Masuma, Satoshi Ōmura and Hiroshi Tomoda
The Journal of Antibiotics 2011 64(7) pp:489-494
Publication Date(Web):May 11, 2011
DOI:10.1038/ja.2011.32
Dinapinone A, a novel biaryl dihydronaphthopyranone, was isolated from the culture broth of Penicillium pinophilum FKI-3864 by solvent extraction, silica gel and ODS column chromatography and HPLC. Dinapinone A showed very potent inhibition of triacylglycerol (TG) synthesis in intact Chinese hamster ovary K1 (CHO-K1) cells with an IC50 value of 0.097 μM. Dinapinone A was found to be a mixture of stereoisomers, resulting in its separation into dinapinones A1 and A2 by HPLC using a C30 reverse-phase column. Dinapinone A1 did not inhibit TG synthesis in CHO-K1 cells even at 12 μM, and dinapinone A2 showed less potent inhibition (IC50; 0.65 μM) than dinapinone A; however, a mixture of isolated dinapinones A1 and A2 (a 1:1 ratio) recovered the potent TG inhibitory activity (IC50; 0.054 μM). A similar effect of dinapinone on TG synthesis in intact Raji cells was also observed.
Co-reporter:Kyosuke Kawamoto, Hiroyuki Yamazaki, Satoshi Ohte, Rokuro Masuma, Ryuji Uchida and Hiroshi Tomoda
The Journal of Antibiotics 2011 64(7) pp:503-508
Publication Date(Web):May 25, 2011
DOI:10.1038/ja.2011.33
Five new monapinones, including a dinapinone monomer, were isolated from the culture broth of the dinapinone-producing Penicillium pinophilum FKI-3864 in a medium modified to contain seawater. The structures of these monapinones were elucidated by various NMR experiments. Monapinones possessed the same dihydronaphthopyranone skeleton as the dinapinones, with different hydroxyalkyl chains: monapinone A was identified as the monomeric portion of the atropisomer dinapinones A1 and A2, and monapinones A and B showed weak inhibition of triacylglycerol (TG) synthesis in intact mammalian cells, whereas the others showed almost no effect on TG synthesis.
Co-reporter:Nobuhiro Koyama, Shigenobu Kojima, Takeo Fukuda, Tohru Nagamitsu, Tadashi Yasuhara, Satoshi O̅mura and Hiroshi Tomoda
Organic Letters 2010 Volume 12(Issue 3) pp:432-435
Publication Date(Web):December 23, 2009
DOI:10.1021/ol902553z
A new fungal metabolite designated calpinactam (1) was isolated from the culture broth of Mortierella alpina FKI-4905, and its structure was elucidated by spectroscopic analyses including NMR experiments. Calpinactam was found to be a hexapeptide with a caprolactam ring at its C-terminal. Its absolute stereochemistry was determined by amino acid analysis and total synthesis. Calpinactam selectively inhibited the growth of mycobacteria among various microorganisms. The MIC values of calpinactam against Mycobacterium smegmatis and M. tuberculosis were 0.78 and 12.5 μg/mL, respectively.
Co-reporter:Hiroyuki Yamazaki, Nobuhiro Koyama, Satoshi O̅mura and Hiroshi Tomoda
Organic Letters 2010 Volume 12(Issue 7) pp:1572-1575
Publication Date(Web):March 11, 2010
DOI:10.1021/ol100298h
New rugulosins B (2) and C (3) were isolated together with known rugulosin (renamed rugulosin A in this paper, 1) from whole culture of Penicillium radicum FKI-3765-2, and their structures were elucidated by NMR spectroscopy. Rugulosins A and C were a homodimer of the same anthraquinone moieties, whereas rugulosin B was a heterodimer of analogous anthraquinone moieties. Rugulosins A to C showed antimicrobial activity against methicillin-resistant Staphylococcus aureus.
Co-reporter:Hiroyuki Yamazaki, Narihiro Ugaki, Daisuke Matsuda and Hiroshi Tomoda
The Journal of Antibiotics 2010 63(6) pp:315-318
Publication Date(Web):April 23, 2010
DOI:10.1038/ja.2010.39
The structure of a new pentacecilide congener, pentacecilide D, produced by Penicillium cecidicola FKI-3765-1 was elucidated by various NMR experiments. The absolute stereochemistry of pentacecilides was elucidated by using the modified Mosher method for pentacecilide C. The inhibitory activity of all pentacecilides against lipid droplet formation and acyl-CoA:cholesterol acyltransferase isozymes was compared.
Co-reporter:Junji Inokoshi, Yoichi Takagi, Ryuji Uchida, Rokuro Masuma, Satoshi Ōmura and Hiroshi Tomoda
The Journal of Antibiotics 2010 63(1) pp:9-16
Publication Date(Web):November 27, 2009
DOI:10.1038/ja.2009.110
Static fermentation of amidepsine-producing fungus Humicola sp. FO-2942 led to the production of six new amidepsines, including a new type of glycosylated congener. Non-glycosylated amidepsine J inhibited both human diacylglycerol acyltransferases 1 (DGAT1) and DGAT2 with the same IC50 value of 40 μM, whereas glycosylated amidepsines F to I showed very weak inhibitory activity against DGAT1 and DGAT2.
Co-reporter:Ryuji Uchida, Masato Iwatsuki, Yong-Pil Kim, Satoshi Ohte, Satoshi Ōmura and Hiroshi Tomoda
The Journal of Antibiotics 2010 63(4) pp:151-155
Publication Date(Web):February 26, 2010
DOI:10.1038/ja.2010.9
The in vivo-mimic assay system using silkworm larvae was used as a screening tool to discover antibiotics against methicillin-resistant Staphylococcus aureus (MRSA). Microbial culture broths were screened in this in vivo-mimic assay system and a culture broth of Streptomyces sp. K04-0144 was selected. New antibiotics, designated nosokomycins A–D, were isolated from the culture broth by HP-20 and ODS column chromatography and HPLC. Nosokomycins inhibited the growth of MRSA with MIC values of 0.125 μg ml−1 using the liquid microdilution method. Furthermore, MRSA-infected silkworms survived when nosokomycin A or B was injected at a dose of 50 μg per larva.
Co-reporter:Ryuji Uchida, Masato Iwatsuki, Yong-Pil Kim, Satoshi Ōmura and Hiroshi Tomoda
The Journal of Antibiotics 2010 63(4) pp:157-163
Publication Date(Web):March 5, 2010
DOI:10.1038/ja.2010.10
The structures of nosokomycins A, B, C and D, new anti-methicillin-resistant Staphylococcus aureus antibiotics produced by Streptomyces sp. K04-0144, were elucidated by spectroscopic studies including various NMR experiments. Nosokomycins A, B, C and D are new members of the moenomycin family consisting of an oligosaccharide moiety, a 2,3-dihydroxypropionic acid and an unusual sesterterpenoid moiety. All nosokomycins lack the cyclopentenone moiety in the oligosaccharide moiety of moenomycin A.
Co-reporter:Nobuhiro Koyama, Shigenobu Kojima, Kenichi Nonaka, Rokuro Masuma, Makoto Matsumoto, Satoshi Ōmura and Hiroshi Tomoda
The Journal of Antibiotics 2010 63(4) pp:183-186
Publication Date(Web):February 26, 2010
DOI:10.1038/ja.2010.14
Calpinactam, a new anti-mycobacterial agent, was isolated from the culture broth of a fungal strain Mortierella alpina FKI-4905 by solvent extraction, octadecyl silane column chromatography and preparative HPLC. Calpinactam was active only against Mycobacteria among various microorganisms, including Gram-positive and Gram-negative bacteria, fungi and yeasts. Calpinactam inhibited the growth of Mycobacterium smegmatis and Mycobacterium tuberculosis with MIC values of 0.78 and 12.5 μg ml−1, respectively.
Co-reporter:Masato Iwatsuki, Yukio Koizumi, Hiroaki Gouda, Shuichi Hirono, Hiroshi Tomoda, Satoshi Ōmura
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 10) pp:2888-2890
Publication Date(Web):15 May 2009
DOI:10.1016/j.bmcl.2009.03.033
C-terminal-lacking fragments of the anti-mycobacterial peptide lariatin A were obtained by hydrolysis using carboxypeptidase P and their anti-mycobacterial activities were evaluated. Lys17 was found to be essential for their antimicrobial activity. A molecular dynamics simulation, with explicit water molecules, helped determine the structural characteristics of Lys17 of lariatin A. The simulation revealed the dynamic formation and deformation of a salt bridge between the Nξ atom of Lys17 and the carboxyl group of C-terminal Pro18, which is deemed to be crucial for the compound’s anti-mycobacterial activity.
Co-reporter:Junji Inokoshi, Kyosuke Kawamoto, Yoichi Takagi, Maki Matsuhama, Satoshi mura and Hiroshi Tomoda
The Journal of Antibiotics 2009 62(1) pp:51-54
Publication Date(Web):January 9, 2009
DOI:10.1038/ja.2008.5
Two isozymes for human acyl-coenzyme A:diacylglycerol acyltransferase (DGAT), DGAT1 and DGAT2, were independently expressed in DGAT-deficient Saccharomyces cerevisiae to establish DGAT1- and DGAT2-S. cerevisiae. The selectivity of DGAT inhibitors of natural origin towards the isozymes was assessed in enzyme assays using the microsomal fractions prepared from DGAT1- and DGAT2-S. cerevisiae. Amidepsines and xanthohumol inhibited DGAT1 and DGAT2 with similar potency, whereas roselipins were found to inhibit DGAT2 selectively.
Co-reporter:Hiroyuki Yamazaki, Satoshi mura and Hiroshi Tomoda
The Journal of Antibiotics 2009 62(4) pp:207-211
Publication Date(Web):March 20, 2009
DOI:10.1038/ja.2009.19
The structures of pentacecilides, new inhibitors of lipid droplet formation in mouse macrophages produced by Penicillium cecidicola FKI-3765-1, were elucidated by spectroscopic studies, including various NMR experiments. Pentacecilides have a common pentacyclic meroterpene core, which contains an aromatic ring and a δ-lactone ring.
Co-reporter:Hiroyuki Yamazaki, Kenichi Nonaka, Rokuro Masuma, Satoshi mura and Hiroshi Tomoda
The Journal of Antibiotics 2009 62(8) pp:431-434
Publication Date(Web):July 24, 2009
DOI:10.1038/ja.2009.69
The fungal strain FKI-3765-2, identified as Penicillium radicum, was found to produce potentiators of imipenem activity against methicillin-resistant Staphylococcus aureus (MRSA). Two new compounds, designated xanthoradones A and B, were isolated from the fermentation broth of the producing strain by solvent extraction, octadecyl silyl column chromatography and preparative HPLC. Xanthoradones A and B potentiated imipenem activity against MRSA by decreasing the MIC value of imipenem from 16 μg ml−1 to 0.060 and 0.030 μg ml−1, respectively.
Co-reporter:Hiroyuki Yamazaki, Satoshi mura and Hiroshi Tomoda
The Journal of Antibiotics 2009 62(8) pp:435-437
Publication Date(Web):July 17, 2009
DOI:10.1038/ja.2009.61
The structures of xanthoradones A and B, new potentiators of imipenem activity against methicillin-resistant Staphylococcus aureus produced by Penicillium radicum FKI-3765-2, were elucidated by spectroscopic studies, including various NMR experiments. These compounds have an asymmetric biaryl skeleton, which contains dihydronaphthopyranone and naphthoquinone moieties.
Co-reporter:Hiroyuki Yamazaki, Kakeru Kobayashi, Daisuke Matsuda, Kenichi Nonaka, Rokuro Masuma, Satoshi mura and Hiroshi Tomoda
The Journal of Antibiotics 2009 62(4) pp:195-200
Publication Date(Web):March 20, 2009
DOI:10.1038/ja.2009.18
New compounds designated pentacecilides A to C were isolated from the fermentation broth of Penicillium cecidicola FKI-3765-1 by solvent extraction, silica gel column chromatography and preparative HPLC. Pentacecilides A and B dose-dependently inhibited lipid droplet formation in mouse macrophages. Furthermore, pentacecilides A and B were found to inhibit the synthesis of cholesteryl ester in mouse macrophages with respective IC50 values of 3.65 and 4.76 μM without any cytotoxic effect, but pentacecilide C showed almost no activity. The study of the mechanism of action strongly suggested that pentacecilides A and B inhibit acyl-CoA: cholesterol acyltransferase activity in macrophages.
Co-reporter:Hiroshi Tomoda and Takayuki Doi
Accounts of Chemical Research 2008 Volume 41(Issue 1) pp:32
Publication Date(Web):September 6, 2007
DOI:10.1021/ar700117b
For discovery of a new type of antiatherosclerotic agents, a cell-based assay of lipid droplet accumulation using primary mouse peritoneal macrophages was conducted as a model of macrophage-derived foam cell accumulation, which occurs in the early stage of atherosclerogenesis. During the screening of microbial metabolites for inhibitors of lipid droplet accumulation, 13-membered cyclodepsipeptides, known beauveriolide I and new beauveriolide III, were isolated from the culture broth of fungal Beauveria sp. FO-6979, a soil isolate, by solvent extraction, ODS column chromatography, silica gel column chromatography, and preparative HPLC. The structure including the absolute stereochemistry of beauveriolide III was elucidated as cyclo-[(3S,4S)-3-hydroxy-4-methyloctanoyl-l-phenylalanyl-l-alanyl-d-alloisoleucyl] by spectral analyses, amino acid analyses, and synthetic methods. Furthermore, the absolute stereochemistry was confirmed by the total synthesis of beauveriolides. Study on the mechanism of action revealed that beauveriolides inhibited macrophage acyl-CoA:cholesterol acyltransferase (ACAT) activity to block the synthesis of cholesteryl ester (CE), leading to a reduction of lipid droplets in macrophages. There are two ACAT isozymes in mammals, ACAT1 and ACAT2. ACAT1 is ubiquitously expressed in most tissues and cells including macrophages, while ACAT2 is expressed predominantly in the liver (hepatocytes) and the intestine (enterocytes). Interestingly, beauveriolides inhibited both ACAT1 and ACAT2 to a similar extent in an enzyme assay that utilized microsomes but inhibited ACAT1 selectively in intact cell-based assays. Beauveriolides proved orally active in both low-density lipoprotein receptor and apolipoprotein E knockout mice, reducing the atheroma lesion of heart and aorta without any side effects such as diarrhea or cytotoxicity to adrenal tissues as observed for many synthetic ACAT inhibitors. To obtain more potent inhibitors, a focused library of beauveriolide analogues was prepared by combinatorial chemistry in which solid-phase assembly of linear depsipeptides was carried out using a 2-chlorotrityl linker, followed by solution-phase cyclization, yielding 104 beauveriolide analogues. Among them, diphenyl derivatives were found to show 10 times more potent inhibition of CE synthesis in macrophages than beauveriolide III. Furthermore, most analogues showed selective ACAT1 inhibition or inhibition of both ACAT1 and ACAT2, but interestingly certain analogues gave selective ACAT2 inhibition. These data indicated that subtle structural differences of the inhibitors could discriminate the active sites of the ACAT1 and ACAT2 isozymes. Efforts of further analogue synthesis would make it possible to obtain highly selective ACAT1/ACAT2 inhibitors.
Co-reporter:Kent Sakai, Satoshi Ohte, Taichi Ohshiro, Daisuke Matsuda, Rokuro Masuma, Lawrence L Rudel and Hiroshi Tomoda
The Journal of Antibiotics 2008 61(9) pp:568-572
Publication Date(Web):2008-09-01
DOI:10.1038/ja.2008.76
Five known fungal metabolites, aurasperone A, aurasperone D, averufanin, flavasperone and sterigmatocystin, were isolated from the culture broths of Aspergillus species as inhibitors of acyl-CoA:cholesterol acyltransferase (ACAT) in the cell-based assay using ACAT1- and ACAT2-expressing CHO cells. These compounds share a similar polycyclic skeleton. Among them, flavasperone and sterigmatocystin, having an angular skeleton, showed selective inhibition toward ACAT2 isozyme, while the others having a linear one had no selectivity in inhibition.
Co-reporter:Takashi Fukuda, Yoko Hasegawa, Yasunari Sakabe, Hiroshi Tomoda and Satoshi mura
The Journal of Antibiotics 2008 61(9) pp:550-555
Publication Date(Web):2008-09-01
DOI:10.1038/ja.2008.73
Two new aromatic alkaloids, designated citrinamides A and B, were isolated from the culture broth of penicillium sp. FKI–1938 by solvent extraction, silica gel column chromatography and HPLC. Their structures were elucidated by spectroscopic analysis, including NMR and amino acid analysis. Citrinamides A and B showed moderate potentiation of miconazole activity against Candida albicans.
Co-reporter:Taichi Ohshiro, Satoshi Ohte, Daisuke Matsuda, Masaki Ohtawa, Tohru Nagamitsu, Toshiaki Sunazuka, Yoshihiro Harigaya, Lawrence L Rudel, Satoshi mura and Hiroshi Tomoda
The Journal of Antibiotics 2008 61(8) pp:503-508
Publication Date(Web):2008-08-01
DOI:10.1038/ja.2008.67
Selectivity of 96 semisynthetic derivatives prepared from fungal pyripyropene A, originally isolated as a potent inhibitor of acyl-CoA:cholesterol acyltransferase (ACAT), toward ACAT1 and ACAT2 isozymes was investigated in the cell-based assay using ACAT1- and ACAT2-expressing CHO cells. Eighteen derivatives including PR-71 (7-O-isocaproyl derivative) showed much more potent ACAT2 inhibition (IC50: 6.0 to 62 nM) than pyripyropene A (IC50: 70 nM). Among them, however, natural pyripyropene A showed the highest selectivity toward ACAT2 with a selectivity index (SI) of > 1000, followed by PR–71 (SI, 667).
Co-reporter:Nobuhiro Koyama, Kakeru Kobayashi, Hiroyuki Yamazaki and Tomoda Hiroshi
The Journal of Antibiotics 2008 61(8) pp:509-514
Publication Date(Web):2008-06-01
DOI:10.1038/ja.2008.68
From a study on the biological activity of fungal stemphones and their derivatives, five derivatives having an O-alkyl moiety at C-11 of stemphone C were found to inhibit lipid droplet accumulation in macrophages without any cytotoxic effect. Among the derivatives, those having O-isopropyl and O-isobutyl were the most potent inhibitors by blocking the synthesis of both cholesteryl ester (CE) and triacylglycerol (TG), the main constituents of lipid droplets in macrophages.
Co-reporter:Daisuke Matsuda, Ichiji Namatame, Taichi Ohshiro, Shun Ishibashi, Satoshi mura and Hiroshi Tomoda
The Journal of Antibiotics 2008 61(5) pp:318-321
Publication Date(Web):2008-05-01
DOI:10.1038/ja.2008.45
As previously reported, triacsin C, a selective inhibitor of acyl-CoA synthetase, inhibited the synthesis of cholesteryl ester and triacylglycerol in mouse peritoneal macrophages, leading to a reduction of lipid droplets. Therefore, the in vivo efficacy was studied. Low-density lipoprotein receptor-knockout (LDLR−/−) mice were fed a high cholesterol diet (0.15%) for two months to measure the atherogenic areas of the hearts and aortas. When triacsin C was orally administered (10 mg/kg/day), the atherosclerotic areas were significantly reduced by 86% in aorta and 36% in hearts. The results strongly suggested that triacsin C shows anti-atherogenic activity by inhibiting acyl-CoA synthetase activity.
Co-reporter:Hiroyuki Yamazaki, Nobuhiro Koyama, Satoshi mura and Hiroshi Tomoda
The Journal of Antibiotics 2008 61(7) pp:426-441
Publication Date(Web):2008-07-01
DOI:10.1038/ja.2008.59
From a further purification study, four new stemphones D to G were isolated along with previously reported stemphones B and C from the culture broth of Aspergillus sp. FKI-2136. Twenty-one derivatives were semisynthetically prepared from stemphones C, E and G. Potentiation of imipenem activity against methicillinresistant Staphylococcus aureus (MRSA) by all the stemphones including natural and semisynthetic ones was compared to study the structure-activity relationships. Derivatives with a free hydroxy or an O-acyl residue having a C2 to C5 carbon length at C-4 held the potentiating activity, but those with a longer acyl residue lost the activity. The presence of an oxo or a free hydroxy residue at C-10 is important for the potentiating activity because introduction of an alkyl or acyl residue at this position resulted in a loss of activity. Among them, stemphone E exhibited the most potent potentiation of imipenem activity against MRSA and the lowest cytotoxic activity against Jurkat cells.
Co-reporter:Masato Iwatsuki, Ryuji Uchida, Hitomi Yoshijima, Hideaki Ui, Kazuro Shiomi, Atsuko Matsumoto, Yoko Takahashi, Akio Abe, Hiroshi Tomoda and Satoshi mura
The Journal of Antibiotics 2008 61(4) pp:222-229
Publication Date(Web):2008-04-01
DOI:10.1038/ja.2008.32
Enteropathogenic Escherichia coli (EPEC) expressing the Type III secretion system (TTSS) induced hemolysis of sheep blood cells. Using this assay, six structurally related compounds designated as guadinomines were isolated as inhibitors of TTSS-induced hemolysis by ion exchange column chromatography and HPLC from the culture broth of Streptomyces sp. K01-0509. Guadinomines A and B showed potent inhibition with IC50 values of 0.02 and 0.007 μg/ml, respectively, guadinomine D showed moderate activity (IC50: 8.5 μg/ml), while guadinomines C1 and C2 and guadinomic acid had no activity.
Co-reporter:Masato Iwatsuki, Ryuji Uchida, Hitomi Yoshijima, Hideaki Ui, Kazuro Shiomi, Yong-Pil Kim, Tomoyasu Hirose, Toshiaki Sunazuka, Akio Abe, Hiroshi Tomoda and Satoshi mura
The Journal of Antibiotics 2008 61(4) pp:230-236
Publication Date(Web):2008-04-01
DOI:10.1038/ja.2008.33
The structures of guadinomines, new inhibitors of a bacterial Type III secretion system produced by Streptomyces sp. K01-0509, were elucidated by spectroscopic studies including various NMR experiments. Guadinomines A, B, C1, C2 and D consist of a carbamoylated cyclic guanidinyl moiety, an alkyl chain moiety and an L-Ala-L-Val moiety in common, while guadinomic acid is a smaller molecule consisting of a carbamoylated cyclic guanidinyl moiety and a hydroxyl hexanoate moiety.
Co-reporter:Atsushi Fukumoto, Yong-Pil Kim, Atsuko Matsumoto, Yoko Takahashi, Kazuro Shiomi, Hiroshi Tomoda and Satoshi Ōmura
The Journal of Antibiotics 2008 61(1) pp:1-6
Publication Date(Web):2008-01-01
DOI:10.1038/ja.2008.101
Cyslabdan, a new potentiator of imipenem activity against methicillin-resistant Staphylococcus aureus, was isolated from the culture broth of Streptomyces sp. K04-0144 by Diaion HP-20 and ODS column chromatographies and preparative HPLC. The structure of cyslabdan was elucidated by spectroscopic analyses including NMR. The compound has a labdane-type diterpene skeleton connecting with an N-acetylcysteine via thioether linkage.
Co-reporter:Atsushi Fukumoto, Yong-Pil Kim, Hideaki Hanaki, Kazuro Shiomi, Hiroshi Tomoda and Satoshi Ōmura
The Journal of Antibiotics 2008 61(1) pp:7-10
Publication Date(Web):2008-01-01
DOI:10.1038/ja.2008.102
Cyslabdan produced by Streptomyces sp. K04-0144 was found to potentiate imipenem activity against methicillin-resistant Staphylococcus aureus (MRSA). The MIC value of imipenem against MRSA was reduced from 16 to 0.015 μg/ml in combination with cyslabdan. Study on anti-MRSA activity of other typical antibiotics in combination with cyslabdan showed that the potentiating activity was limited to β-lactam antibiotics. Furthermore, among β-lactam antibiotics, the activity of carbapenems was most remarkably poteintiated by cyslabdan.
Co-reporter:Masato Iwatsuki, Ryuji Uchida, Yoichi Takakusagi, Atsuko Matsumoto, Cheng-Lin Jiang, Yoko Takahashi, Masayoshi Arai, Susumu Kobayashi, Makoto Matsumoto, Junji Inokoshi, Hiroshi Tomoda and Satoshi mura
The Journal of Antibiotics 2007 60(6) pp:357-363
Publication Date(Web):2007-06-01
DOI:10.1038/ja.2007.48
Two anti-mycobacterial peptides with a lasso structure, named lariatins A and B, were separated by HP-20 and ODS column chromatographies and purified by HPLC from the culture broth of Rhodococcus jostii K01-B0171, which was isolated from soil aggregates collected in Yunnan, China. Lariains A and B showed growth inhibition against Mycobacterium smegmatis with MIC values of 3.13 and 6.25 g/ml in agar dilution method, respectively. Furthermore, lariatin A inhibited the growth of Mycobacterium tuberculosis with an MIC of 0.39 g/ml in liquid microdilution method.
Co-reporter:Taichi Ohshiro, Lawrence L Rudel, Satoshi mura and Hiroshi Tomoda
The Journal of Antibiotics 2007 60(1) pp:43-51
Publication Date(Web):2007-01-01
DOI:10.1038/ja.2007.6
The selectivity of microbial inhibitors of acyl-CoA : cholesterol acyltransferase (ACAT) toward the two isozymes, ACAT1 and ACAT2, was assessed in cell-based assays. Purpactin A (IC50 values of ACAT1 vs. IC50 values of ACAT2; 2.5 M vs. 1.5 M), terpendole C (10 M vs. 10 M), glisoprenin A (4.3 M vs. 10 M), spylidone (25 M vs. 5.0 M) and synthetic CL-283,546 (0.1 M vs. 0.09 M) inhibited ACAT1 and ACAT2 to similar extents. Beauveriolides I (0.6 M vs. 20 M) and III (0.9 M vs. >20 M) inhibited ACAT1 rather selectively, while pyripyropenes A (>80 M vs. 0.07 M), B (48 M vs. 2.0 M), C (32 M vs. 0.36 M) and D (38 M vs. 1.5 M) showed selective inhibition against ACAT2. In particular, pyripyropene A was found to be the most selective ACAT2 inhibitor with a selective index of more than 1,000.
Co-reporter:Ryuji Uchida, Yong-Pil Kim, Ichiji Namatame, Hiroshi Tomoda and Satoshi mura
The Journal of Antibiotics 2006 59(2) pp:93-97
Publication Date(Web):
DOI:10.1038/ja.2006.13
Sespendole was isolated as an inhibitor of lipid droplet formation in macrophages from the culture broth of a fungal strain Pseudobotrytis terrestris FKA-25. The compound inhibited the synthesis of cholesteryl ester and triacylglycerol by mouse macrophages with IC50 values of 4.0 and 3.2 M, respectively.
Co-reporter:Ryuji Uchida, Rie Imasato, Yuichi Yamaguchi, Rokuro Masuma, Kazuro Shiomi, Hiroshi Tomoda and Satoshi mura
The Journal of Antibiotics 2006 59(10) pp:646-651
Publication Date(Web):
DOI:10.1038/ja.2006.86
New nine insecticidal antibiotics designated yaequinolones were isolated from the culture broth of the fungal strain Penicillium sp. FKI-2140 by solvent extraction, centrifugal partition chromatography and HPLC. Yaequinolones showed growth inhibitory activity against brine shrimp (Artemia salina). Among them, yaequinolone F has the most potent activity with MIC value of 0.19 g/ml.
Co-reporter:Ryuji Uchida, Rie Imasato, Hiroshi Tomoda and Satoshi mura
The Journal of Antibiotics 2006 59(10) pp:652-658
Publication Date(Web):
DOI:10.1038/ja.2006.87
The structure and relative stereochemistry of yaequinolones, fungal insecticidal antibiotics, were elucidated by spectroscopic studies, including NMR spectral analyses. Yaequinolones possess a p-methoxyphenylquinolinone skeleton modified with different isoprenyl-derived side chains.
Co-reporter:Takashi Fukuda, Yoko Hasegawa, Keiichi Hagimori, Yuichi Yamaguchi, Rokuro Masuma, Hiroshi Tomoda and Satoshi mura
The Journal of Antibiotics 2006 59(8) pp:480-485
Publication Date(Web):
DOI:10.1038/ja.2006.67
Two new furopyrrols, designated tensidols A and B, were isolated from the culture broth of Aspergillus niger FKI-2342 by solvent extraction, silica gel column chromatography and HPLC. Their structures were elucidated and shown to have the common skeleton of 6-benzyl-6H-furo[2,3-b]pyrrole. Tensidols A and B potentiated miconazole activity against Candida albicans. Tensidols also showed moderate antimicrobial activity only against Pyricularia oryzae.
Co-reporter:Ryuji Uchida, Yong-Pil Kim, Tohru Nagamitsu, Hiroshi Tomoda and Satoshi mura
The Journal of Antibiotics 2006 59(6) pp:338-344
Publication Date(Web):
DOI:10.1038/ja.2006.47
A new fungal metabolite named sespendole was isolated as an inhibitor of lipid droplet synthesis in mouse macrophages from the culture broth of the fungal strain Pseudobotrytis terrestris FKA-25. The structure and stereochemistry of sespendole were elucidated by spectroscopic studies including various NMR spectral analyses, exciton chirality experiments and the modified Mosher method. Sespendole was found to possess a new indolosesquiterpene skeleton modified with two isoprenes.
Co-reporter:Ryuji Uchida, Hiroshi Tomoda and Satoshi mura
The Journal of Antibiotics 2006 59(5) pp:298-302
Publication Date(Web):
DOI:10.1038/ja.2006.42
Sespendole is the first reported fungal metabolite having an indolosesquiterpene core structure. The biosynthesis of sespendole was studied here by feeding experiments with [13C]acetate, [15N]anthranilic acid and [13C]tryptophan. The data suggested that a farnesyl residue derived from the mevalonate pathway and an anthranilate-derived indole-3-glycerol phosphate residue are condensed, and then cyclization occurs along with rearrangement to form the indolosesquiterpene core.
Co-reporter:Hiroshi Tomoda, Satoshi Ōmura
Pharmacology & Therapeutics (September 2007) Volume 115(Issue 3) pp:375-389
Publication Date(Web):1 September 2007
DOI:10.1016/j.pharmthera.2007.05.008
Diacylglycerol acyltransferase (DGAT) and acyl-CoA: cholesterol acyltransferase (ACAT) are the enzymes that catalyze the final reactions of triacylgycerol (TG) and cholesteryl ester (CE) synthesis, and accumulation of TG and CE in adipocytes and arteries causes obesity and atherosclerosis, respectively. Therefore, DGAT and ACAT have been viewed as potential therapeutic targets for these diseases. From the screening program for DGAT inhibitors, new compounds were discovered from fungal and plant extracts, and are expected to provide leads for drug development. From the screening programs for ACAT inhibitors and lipid droplet synthesis inhibitors, new compounds with chemical structures different from those of known synthetic inhibitors were discovered from the cultures of fungal and actinomycete strains. Among them, fungal beauveriolide III rather selectively inhibited ACAT1 isozyme, while fungal pyripyropene A was found to be a highly selective inhibitor of ACAT2 isozyme. Both inhibitors proved orally active in in vivo models. Furthermore, a library of beauveriolide and pyripyropene analogs was prepared by combinatorial and semisynthetic methods, respectively. The future prospects of these inhibitors are discussed.
Co-reporter:Nobuhiro Koyama, Hirofumi Sato, Hiroshi Tomoda
Acta Pharmaceutica Sinica B (November 2015) Volume 5(Issue 6) pp:564-568
Publication Date(Web):November 2015
DOI:10.1016/j.apsb.2015.07.010
Co-reporter:Ryuji Uchida, Mio Kawaguchi, Noriko Sato, Hiroshi Tomoda
Acta Pharmaceutica Sinica B (May 2013) Volume 3(Issue 3) pp:
Publication Date(Web):1 May 2013
DOI:10.1016/j.apsb.2013.04.005
Monapinones A (1) to E (5), half parts of dinapinones, were produced by fermentation of Talaromyces pinophilus FKI-3864 in seawater-containing medium and have a common dihydronaphthopyranone skeleton with a different long alkyl chain. The relative stereochemistries of 3–5 were elucidated by various NMR experiments including analysis of 1H NMR coupling constants, ROESY and the dihedral angles. The absolute stereochemistries of 3–5 at C-3 were determined by the circular dichroism spectra in comparison to the data of (R)- and (S)-semivioxanthins (6 and 7). Accordingly, total absolute stereochemistries of 3–5 were concluded to be 3S,13R,15R,17R,19R,3S,13R,15R,17R and 3S,13R,15R, respectively.Monapinones A (1)–E (5), half parts of dinapinones, were produced by fermentation of Talaromyces pinophilus FKI-3864 in seawater-containing medium and have a common dihydronaphthopyranone skeleton with a different long alkyl chain. Total absolute stereochemistries of 3–5 were concluded to be 3S, 13R, 15R, 17R, 19R, 3S, 13R, 15R, 17R and 3S, 13R, 15R, respectively. Download full-size image
Co-reporter:Nobuhiro Koyama, Yuriko Tokura, Yoko Takahashi, Hiroshi Tomoda
Acta Pharmaceutica Sinica B (December 2011) Volume 1(Issue 4) pp:236-239
Publication Date(Web):December 2011
DOI:10.1016/j.apsb.2011.10.008
Co-reporter:Ryuji Uchida, Seiko Ishikawa, Hiroshi Tomoda
Acta Pharmaceutica Sinica B (April 2014) Volume 4(Issue 2) pp:
Publication Date(Web):1 April 2014
DOI:10.1016/j.apsb.2013.12.008
2-Hydroxytyrosol (2-HT), originally reported as a synthetic compound, was isolated for the first time as a fungal metabolite. 2-HT was found to inhibit mushroom tyrosinase with an IC50 value of 13.0 µmol/L. Furthermore, 2-HT dose-dependently inhibited tyrosinase activity (IC50, 32.5 µmol/L) in the cell-free extract of B16 melanoma cells and α-melanocyte stimulating hormone (α-MSH)-stimulated melanin formation in intact B16 melanoma cells.2-Hydroxytyrosol (2-HT), was found to inhibit mushroom tyrosinase activity (IC50, 32.5 µmol/L) in the cell-free extract of B16 melanoma cells and α-melanocyte stimulating hormone (α-MSH)-stimulated melanin formation in intact B16 melanoma cells.Download full-size image
Co-reporter:Keisuke Kobayashi, Taichi Ohshiro, Hiroshi Tomoda, Feng Yin, Hai-Lei Cui, Pandurang V. Chouthaiwale, Fujie Tanaka
Bioorganic & Medicinal Chemistry Letters (15 December 2016) Volume 26(Issue 24) pp:
Publication Date(Web):15 December 2016
DOI:10.1016/j.bmcl.2016.11.008
Synthesis of new functionalized molecules and identification of biofunctional molecules can lead to the development of therapeutic leads and molecular tools for biomedical research. We have recently reported oxa-hetero-Diels-Alder reactions of enones with isatins to provide functionalized spirooxindole tetrahydropyran derivatives. Twenty-one compounds from the spirooxindole tetrahydropyran derivatives and related molecules were screened for inhibition of sterol O-acyltransferase (SOAT) isozymes SOAT1 and SOAT2. Three racemic derivatives inhibited the SOAT2 isozyme with three-fold or better selectivity for SOAT2 than for SOAT1. The enantiomerically enriched forms of the most efficient racemic inhibitor of SOAT2 were further evaluated; one enantiomer inhibited SOAT2 with an IC50 of 1.5 μM and was 10-fold more selective for SOAT2 than SOAT1.
Co-reporter:Takashi Fukuda, Ryuji Uchida, Hiroyo Inoue, Satoshi Ohte, Hiroyuki Yamazaki, Daisuke Matsuda, Takenobu Katagiri, Hiroshi Tomoda
Acta Pharmaceutica Sinica B (10 February 2012) Volume 2(Issue 1) pp:23-27
Publication Date(Web):10 February 2012
DOI:10.1016/j.apsb.2011.12.011

![1,7-BIS[(Z)-BUT-2-EN-2-YL]-3,9-DIHYDROXY-4,10-DIMETHYLBENZO[B][1,4]BENZODIOXEPIN-6-ONE](http://img.cochemist.com/ccimg/140000/139959-71-0.png)
![1,7-BIS[(Z)-BUT-2-EN-2-YL]-3,9-DIHYDROXY-4,10-DIMETHYLBENZO[B][1,4]BENZODIOXEPIN-6-ONE](http://img.cochemist.com/ccimg/140000/139959-71-0_b.png)
![(6R,7R)-7-AMIMO-8-OXO-3-(1-PROPENYL)-5-THIA-1-azabicyclo[4.2.0]OCT-2-ene-2-carboxylic acid monohydrate](http://img.cochemist.com/ccimg/74500/74431-23-5.png)
![(6R,7R)-7-AMIMO-8-OXO-3-(1-PROPENYL)-5-THIA-1-azabicyclo[4.2.0]OCT-2-ene-2-carboxylic acid monohydrate](http://img.cochemist.com/ccimg/74500/74431-23-5_b.png)
![(8,8'-BI-1H-NAPHTHO[2,3-C]PYRAN)-3,3'-DIACETIC ACID, 3,3',4,4'-TETRAHYDRO-9,9',10,10'-TETRAHYDROXY-7,7'-DIMETHOXY-1,1'-DIOXO-, DIMETHYL ESTER](http://img.cochemist.com/ccimg/39300/39277-41-3.png)
![(8,8'-BI-1H-NAPHTHO[2,3-C]PYRAN)-3,3'-DIACETIC ACID, 3,3',4,4'-TETRAHYDRO-9,9',10,10'-TETRAHYDROXY-7,7'-DIMETHOXY-1,1'-DIOXO-, DIMETHYL ESTER](http://img.cochemist.com/ccimg/39300/39277-41-3_b.png)

























![Pyrazino[1'',2'':1,5;4'',5'':1',5']dipyrrolo[2,3-b:2',3'-b']diindole-7,15(5H,7aH)-dione,8a,16a-bis(1,1-dimethyl-2-propen-1-yl)-5a,8,8a,13,13a,15a,16,16a-octahydro-,(5aS,7aS,8aR,13aS,15aS,16aR)-](http://img.cochemist.com/ccimg/88400/88360-87-6.png)
![Pyrazino[1'',2'':1,5;4'',5'':1',5']dipyrrolo[2,3-b:2',3'-b']diindole-7,15(5H,7aH)-dione,8a,16a-bis(1,1-dimethyl-2-propen-1-yl)-5a,8,8a,13,13a,15a,16,16a-octahydro-,(5aS,7aS,8aR,13aS,15aS,16aR)-](http://img.cochemist.com/ccimg/88400/88360-87-6_b.png)



![methyl (2R,4aR,4bS,10aS,12aR)-6,10b-dihydroxy-2,4b,7,7,10a,12a-hexamethyl-12-methylidene-1,4,5,8-tetraoxo-1,4,4a,4b,5,7,8,9,10,10a,10b,11,12,12a-tetradecahydro-2H-naphtho[1,2-h]isochromene-2-carboxylate](http://img.cochemist.com/ccimg/72000/71911-90-5.png)
![methyl (2R,4aR,4bS,10aS,12aR)-6,10b-dihydroxy-2,4b,7,7,10a,12a-hexamethyl-12-methylidene-1,4,5,8-tetraoxo-1,4,4a,4b,5,7,8,9,10,10a,10b,11,12,12a-tetradecahydro-2H-naphtho[1,2-h]isochromene-2-carboxylate](http://img.cochemist.com/ccimg/72000/71911-90-5_b.png)
![4H,11H-Naphtho[2,1-b]pyrano[3,4-e]pyran-1,11(5H)-dione,4a,6,6a,12,12a,12b-hexahydro-4a,12a-dihydroxy-4,4,6a,12b-tetramethyl-9-(3,4,5-trimethoxyphenyl)-,(4aR,6aR,12aS,12bS)-](http://img.cochemist.com/ccimg/70500/70407-20-4.png)
![4H,11H-Naphtho[2,1-b]pyrano[3,4-e]pyran-1,11(5H)-dione,4a,6,6a,12,12a,12b-hexahydro-4a,12a-dihydroxy-4,4,6a,12b-tetramethyl-9-(3,4,5-trimethoxyphenyl)-,(4aR,6aR,12aS,12bS)-](http://img.cochemist.com/ccimg/70500/70407-20-4_b.png)



![Spiro[2-benzoxepin-9(3H),2'-oxirane]-4-carboxylicacid, 1,5a,6,7,8,9a-hexahydro-6-(1-methylethyl)-1-oxo-, (2'S,5aS,6R,9aS)-](http://img.cochemist.com/ccimg/57800/57710-57-3.png)
![Spiro[2-benzoxepin-9(3H),2'-oxirane]-4-carboxylicacid, 1,5a,6,7,8,9a-hexahydro-6-(1-methylethyl)-1-oxo-, (2'S,5aS,6R,9aS)-](http://img.cochemist.com/ccimg/57800/57710-57-3_b.png)

![5-acetyl-14a-(1,1,-dimethyl-allyl)-5a,13a,14,14a-tetrahydro-5H,12H-benzo[5',6'][1,4]diazepino[1',2':1,5]pyrrolo[2,3-b]indole-7,13-dione](http://img.cochemist.com/ccimg/42300/42230-55-7.png)
![5-acetyl-14a-(1,1,-dimethyl-allyl)-5a,13a,14,14a-tetrahydro-5H,12H-benzo[5',6'][1,4]diazepino[1',2':1,5]pyrrolo[2,3-b]indole-7,13-dione](http://img.cochemist.com/ccimg/42300/42230-55-7_b.png)















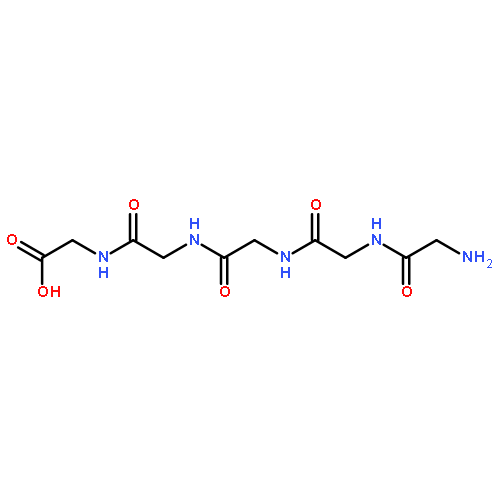
![Diphosphoric acid,P-[(2E,6E,10E)-3,7,11,15-tetramethyl-2,6,10,14-hexadecatetraen-1-yl] ester](http://img.cochemist.com/ccimg/6700/6699-20-3.png)
![Diphosphoric acid,P-[(2E,6E,10E)-3,7,11,15-tetramethyl-2,6,10,14-hexadecatetraen-1-yl] ester](http://img.cochemist.com/ccimg/6700/6699-20-3_b.png)








![Bicyclo[4.1.0]heptane-3,4-diol,3,7,7-trimethyl-, (1S,3S,4S,6R)-](http://img.cochemist.com/ccimg/2200/2140-46-7.png)
![Bicyclo[4.1.0]heptane-3,4-diol,3,7,7-trimethyl-, (1S,3S,4S,6R)-](http://img.cochemist.com/ccimg/2200/2140-46-7_b.png)


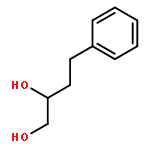
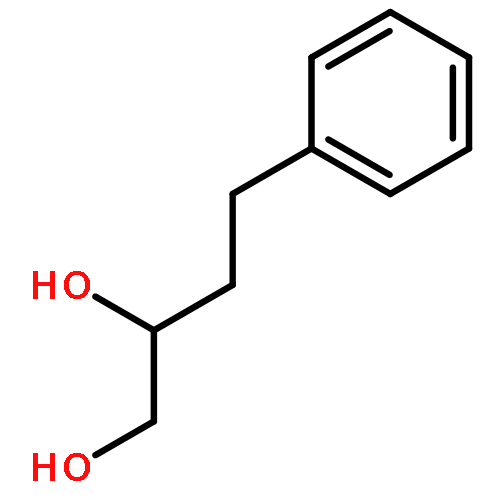
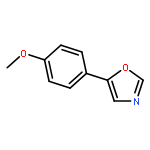






![4-Thia-1-azabicyclo[3.2.0]heptane-2-carboxylicacid, 6-[(2,6-dimethoxybenzoyl)amino]-3,3-dimethyl-7-oxo-, (2S,5R,6R)-](http://img.cochemist.com/ccimg/100/61-32-5.png)
![4-Thia-1-azabicyclo[3.2.0]heptane-2-carboxylicacid, 6-[(2,6-dimethoxybenzoyl)amino]-3,3-dimethyl-7-oxo-, (2S,5R,6R)-](http://img.cochemist.com/ccimg/100/61-32-5_b.png)


![2-Butenedioic acid(2E)-,1-[(2R,4R,5S,6R)-tetrahydro-2-hydroxy-2-[(1S,2R,3S)-2-hydroxy-3-[(2R,3S,4E,6E,9S,10S,11R,12E,14Z)-10-hydroxy-3,15-dimethoxy-7,9,11,13-tetramethyl-16-oxooxacyclohexadeca-4,6,12,14-tetraen-2-yl]-1-methylbutyl]-5-methyl-6-(1-methylethyl)-2H-pyran-4-yl]ester](http://img.cochemist.com/ccimg/89000/88979-61-7.png)
![2-Butenedioic acid(2E)-,1-[(2R,4R,5S,6R)-tetrahydro-2-hydroxy-2-[(1S,2R,3S)-2-hydroxy-3-[(2R,3S,4E,6E,9S,10S,11R,12E,14Z)-10-hydroxy-3,15-dimethoxy-7,9,11,13-tetramethyl-16-oxooxacyclohexadeca-4,6,12,14-tetraen-2-yl]-1-methylbutyl]-5-methyl-6-(1-methylethyl)-2H-pyran-4-yl]ester](http://img.cochemist.com/ccimg/89000/88979-61-7_b.png)


![2H,11H-Naphtho[2,1-b]pyrano[3,4-e]pyran-11-one,3,6-bis(acetyloxy)-4-[(acetyloxy)methyl]-1,3,4,4a,5,6,6a,12,12a,12b-decahydro-12-hydroxy-4,6a,12b-trimethyl-9-(3-pyridinyl)-,(3S,4R,4aR,6S,6aS,12R,12aS,12bS)-](http://img.cochemist.com/ccimg/147500/147444-03-9.png)
![2H,11H-Naphtho[2,1-b]pyrano[3,4-e]pyran-11-one,3,6-bis(acetyloxy)-4-[(acetyloxy)methyl]-1,3,4,4a,5,6,6a,12,12a,12b-decahydro-12-hydroxy-4,6a,12b-trimethyl-9-(3-pyridinyl)-,(3S,4R,4aR,6S,6aS,12R,12aS,12bS)-](http://img.cochemist.com/ccimg/147500/147444-03-9_b.png)