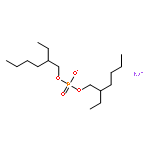
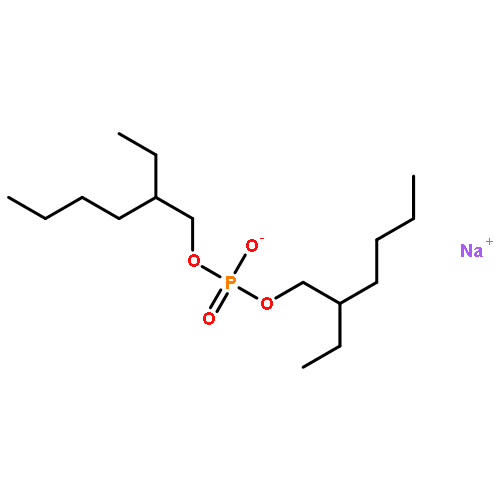
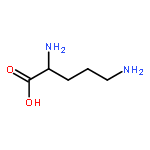
Co-reporter:Xiao-Fei WANG, Shou-Fang JIANG, Wei-Bing ZHANG, Ling-Yi ZHANG, ... He-Qing SHEN
Chinese Journal of Analytical Chemistry 2017 Volume 45, Issue 5(Volume 45, Issue 5) pp:
Publication Date(Web):1 May 2017
DOI:10.1016/S1872-2040(17)61011-9
Airborne fine particulate matter (PM2.5) pollution is a serious environmental problem, thus it is very important to study the toxicity effect and mechanism of PM2.5. In this study, liquid chromatography-mass spectrometry (LC-MS)-based metabolomics technique was used to investigate the metabolic disruption effect and reproductive toxicity mechanism of PM2.5 on rat testis. The profile of aqueous and organic testis metabolic extracts were acquired, and then analyzed by partial least square-discriminant analysis and nonparametric test. The results showed that control group and treatment group were clearly discriminated in the scoring plot of partial least squares discriminant analysis (PLS-DA) models, indicating PM2.5 treatment induced significant difference in testis metabolome, a total of 56 differential metabolites were identified, and further pathway analysis suggested that PM2.5 exposure induced amino acid and nucleotide metabolism disorder, steroid hormone metabolism imbalance and abnormal lipid metabolism. These important pathways may be the key molecular evens in the PM2.5 reproductive toxicity.Airborne fine particulate matter (PM2.5) is a serious environmental problem. In this study, a liquid chromatography-mass spectrometry-based metabolomics technique was adopted to investigate the metabolic disruption effect and reproductive toxicity mechanism of PM2.5. The results showed that PM2.5 treatment induced significant alteration in testis metabolome, and pathway analysis suggested that PM2.5 treatment induced amino acid and nucleotide metabolism disorder, steroid hormone metabolism imbalance and abnormal lipid metabolism. These important pathways may be the potential molecular events of PM2.5 reproductive toxicity.Download high-res image (100KB)Download full-size image
Co-reporter:Jie Zhang, Liangpo Liu, Xiaofei Wang, Qingyu Huang, Meiping Tian, and Heqing Shen
Environmental Science & Technology 2016 Volume 50(Issue 11) pp:5953-5960
Publication Date(Web):May 3, 2016
DOI:10.1021/acs.est.6b00034
The general population is exposed to phthalates through various sources and routes. Integration of omics data and epidemiological data is a key step toward directly linking phthalate biomonitoring data with biological response. Urine metabolomics is a powerful tool to identify exposure biomarkers and delineate the modes of action of environmental stressors. The objectives of this study are to investigate the association between low-level environmental phthalate exposure and urine metabolome alteration in male population, and to unveil the metabolic pathways involved in the mechanisms of phthalate toxicity. In this retrospective cross-sectional study, we studied the urine metabolomic profiles of 364 male subjects exposed to low-level environmental phthalates. Di(2-ethylhexyl) phthalate (DEHP) and dibutyl phthalate (DBP) are the most widely used phthalates. ∑DEHP and MBP (the major metabolite of DBP) were associated with significant alteration of global urine metabolome in the male population. We observed significant increase in the levels of acetylneuraminic acid, carnitine C8:1, carnitine C18:0, cystine, phenylglycine, phenylpyruvic acid and glutamylphenylalanine; and meanwhile, decrease in the levels of carnitine C16:2, diacetylspermine, alanine, taurine, tryptophan, ornithine, methylglutaconic acid, hydroxyl-PEG2 and keto-PGE2 in high exposure group. The observations indicated that low-level environmental phthalate exposure associated with increased oxidative stress and fatty acid oxidation and decreased prostaglandin metabolism. Urea cycle, tryptophan and phenylalanine metabolism disruption was also observed. The urine metabolome disruption effects associated with ∑DEHP and MBP were similar, but not identical. The multibiomarker models presented AUC values of 0.845 and 0.834 for ∑DEHP and MBP, respectively. The predictive accuracy rates of established models were 81% for ΣDEHP and 73% for MBP. Our results suggest that low-level environmental phthalate exposure associates with urine metabolome disruption in male population, providing new insight into the early molecular events of phthalate exposure.
Co-reporter:Xiaoxue Wang, Xiaoli Mu, Jie Zhang, Qingyu Huang, Ambreen Alamdar, Meiping Tian, Liangpo Liu and Heqing Shen
Metallomics 2015 vol. 7(Issue 3) pp:544-552
Publication Date(Web):11 Feb 2015
DOI:10.1039/C5MT00002E
Chronic arsenic exposure through drinking water threatens public health worldwide. Although its multiorgan toxicity has been reported, the impact of chronic arsenic exposure on the metabolic network remains obscure. In this study, male Sprague Dawley rats were exposed to 0.5, 2 or 10 ppm sodium arsenite for three months. An ultra-high performance liquid chromatography/mass spectrometry based metabolomics approach was utilized to unveil the global metabolic response to chronic arsenic exposure in rats. Distinct serum metabolome profiles were found to be associated with the doses. Eighteen differential metabolites were identified, and most of them showed dose-dependent responses to arsenic exposure. Metabolic abnormalities mainly involved lipid metabolism and amino acid metabolism. The metabolic alterations were further confirmed by hepatic gene expression. Expressions of cpt2, lcat, cact, crot and mtr were significantly elevated in high dose groups. This study provides novel evidence to support the association between arsenic exposure and metabolic disruption, and it contributes to understanding the mechanism of chronic arsenic toxicity.
Co-reporter:Xiaoxue Wang, Jie Zhang, Qingyu Huang, Ambreen Alamdar, Meiping Tian, Liangpo Liu and Heqing Shen
Molecular BioSystems 2015 vol. 11(Issue 3) pp:753-759
Publication Date(Web):26 Nov 2014
DOI:10.1039/C4MB00565A
Benzo(a)pyrene [B(a)P] is ubiquitous in the environment. Although multiple toxicities have been reported for B(a)P, the impact of exposure to this chemical on metabolic networks remains obscure. In this study, a metabolomics approach based on ultra-high-performance liquid chromatography/mass spectrometry was used to investigate the disruption of global serum metabolic profiles in rats caused by exposure to B(a)P. Sprague–Dawley rats were treated with oral doses of 10, 100 and 1000 μg kg−1 B(a)P for 32 consecutive days. Distinct serum metabolomic profiles were associated with these doses. Twelve metabolites were identified as potential biomarkers and indicated that exposure to B(a)P disrupted both global amino acid metabolism and lipid metabolism, especially phospholipid and sphingolipid metabolism. Serum levels of lysophosphatidylcholines showed dose-dependent decreases, whereas serum levels of sphingomyelins showed dose-dependent increases. The expressions of some key genes involved in these pathways were also investigated. Expressions of enpp2, sms and smpd were significantly altered by exposure to high doses of B(a)P. Metabolic biomarkers were more sensitive than the corresponding gene expression for exposure to B(a)P. The findings of this study suggest potential novel mechanisms for the identified metabolic pathways.
Co-reporter:Qingyu Huang;Siyuan Peng;Miaomiao Du;Sawyen Ow;Hai Pu;Chensong Pan;Heqing Shen
Journal of Applied Toxicology 2014 Volume 34( Issue 12) pp:1342-1351
Publication Date(Web):
DOI:10.1002/jat.2963
ABSTRACT
Perfluorooctane sulfonate (PFOS) is one of the most commonly used perfluorinated compounds, whose environmental exposure has been associated with a number of adverse health outcomes. However, the molecular mechanisms involved in PFOS toxicity are still not well elucidated. In the present study, we applied iTRAQ labeling quantitative proteomic technology to investigate the differential protein expression profiles of non-tumor human hepatic cells (L-02) exposed to PFOS. A total of 18 proteins were differentially expressed in a dose-dependent manner in PFOS-treated cells versus the control. Among these, 11 proteins were up-regulated and 7 were down-regulated. Gene ontology analysis indicated that PFOS would exert toxic effects on L-02 cells by affecting multiple biological processes, including protein biosynthesis and degradation, mRNA processing and splicing, transcription, signal transduction and transport. Furthermore, the proteomic results especially proposed that the inhibition of HNRNPC, HUWE1 and UBQLN1, as well as the induction of PAF1 is involved in the activation of the p53 and c-myc signaling pathways, which then trigger the apoptotic process in L-02 cells exposed to PFOS. Overall, these data will aid our understanding of the mechanisms responsible for PFOS-mediated hepatotoxicity, and develop useful biomarkers for monitoring and evaluating PFOS contamination in the environment. Copyright © 2013 John Wiley & Sons, Ltd.
Co-reporter:Qingyu Huang;Siyuan Peng;Meiping Tian;Jinsheng Chen ;Heqing Shen
Journal of Applied Toxicology 2014 Volume 34( Issue 6) pp:675-687
Publication Date(Web):
DOI:10.1002/jat.2910
ABSTRACT
Exposure to airborne particulate matter (PM)2.5, a PM with aerodynamic diameter of less than 2.5 µm, is known to be associated with a variety of adverse health effects. However, the molecular mechanisms involved in fine PM toxicity are still not well characterized. The present study aims to provide new insights into the cytotoxicity of PM2.5 on human lung epithelial cells (A549) at the proteomic level. Two-dimensional difference gel electrophoresis revealed a total of 27 protein spots, whose abundance were significantly altered in A549 cells exposed to water-soluble PM2.5 extracts (WSPE). Among these, 12 spots were upregulated while 15 were downregulated. Twenty-two proteins were further identified by matrix-assisted laser desorption/ionization time-of-flight tandem mass/mass spectrometry and database search. The results revealed that oxidative stress, metabolic disturbance, dysregulation of signal transduction, aberrant protein synthesis and degradation, as well as cytoskeleton disorganization are major factors contributing to WSPE-mediated toxicity in human lung cells. It is further proposed that induction of apoptosis through p53, c-Myc and p21 pathways may be one of the key toxicological events occurred in A549 cells under WSPE stress. The data obtained here will aid our understanding of the toxic mechanisms related to PM2.5, and develop useful biomarkers indicative of inhalable PM2.5 exposure. Copyright © 2013 John Wiley & Sons, Ltd.
Co-reporter:Siyuan Peng, Lijuan Yan, Jie Zhang, Zhanlin Wang, Meiping Tian, Heqing Shen
Journal of Pharmaceutical and Biomedical Analysis 2013 Volume 86() pp:56-64
Publication Date(Web):December 2013
DOI:10.1016/j.jpba.2013.07.014
•A multi-omics approach was used to investigate PFOA-induced hepatotoxicity.•Fifteen potential biomarkers were identified on metabolic level.•The mitochondrial carnitine metabolism was proved to be accelerated.•The cholesterol biosynthesis was directly confirmed to be up-regulated.•Amino acid metabolism and tricarboxylic acid cycle might also be disturbed.Perfluorooctanoic acid (PFOA) is one of the most representative perfluorinated compounds and liver is the major organ where PFOA is accumulated. Although the multiple toxicities had been reported, its toxicological profile remained unclear. In this study, a systems toxicology strategy integrating liquid chromatography/mass spectrometry-based metabonomics and transcriptomics analyses was applied for the first time to investigate the effects of PFOA on a representative Chinese normal human liver cell line L-02, with focusing on the metabolic disturbance. Fifteen potential biomarkers were identified on metabolic level and most observations were consistent with the altered levels of gene expression. Our results showed that PFOA induced the perturbations in various metabolic processes in L-02 cells, especially lipid metabolism-related pathways. The up-stream mitochondrial carnitine metabolism was proved to be influenced by PFOA treatment. The specific transformation from carnitine to acylcarnitines, which showed a dose-dependent effect, and the expression level of key genes involved in this pathway were observed to be altered correspondingly. Furthermore, the down-stream cholesterol biosynthesis was directly confirmed to be up-regulated by both increased cholesterol content and elevated expression level of key genes. The PFOA-induced lipid metabolism-related effects in L-02 cells started from the fatty acid catabolism in cytosol, fluctuated to the processes in mitochondria, extended to the cholesterol biosynthesis. Many other metabolic pathways like amino acid metabolism and tricarboxylic acid cycle might also be disturbed. The findings obtained from the systems biological research provide more details about metabolic disorders induced by PFOA in human liver.
Co-reporter:Li-Juan YAN, Jie ZHANG, Chen-Song PAN, Li-Yi LIN, Xin-Yi ZHANG, He-Qing SHEN
Chinese Journal of Analytical Chemistry 2013 Volume 41(Issue 1) pp:31-35
Publication Date(Web):January 2013
DOI:10.1016/S1872-2040(13)60621-0
A high throughput screening method was developed for the simultaneous analysis of twenty representative tranquilizers in dairy products using the combination of ultra-performance liquid chromatography-high resolution time-of-flight mass spectrometry and screening database built with Target Analysis software. The protein and fat in the sample was removed using a two-step precipitation of acetonitrile and acidic acetonitrile, and the supernatant was combined and concentrated with Speedvac concentrator. The chromatographic separation was performed on an ACQUITY BEH column with gradient elution using 0.1% formic acid and acetonitrile as mobile phase at a flow rate of 0.3 mL min−1. The monitoring of tranquilizers was achieved by time-of-flight mass spectrometry under positive ionization mode in 9 min. The quantification was performed with matrix-matching method. The linear ranges were 1–500 μg L−1 or 5–1000 μg L−1. The LODs and LOQs were 0.3–1.5 μg L−1 and 1–5 μg L−1, respectively. At 5 and 50μg L−1 spiked levels, the average recoveries were 76.1%–108.2% with relative standard deviations of 2.5%–9.0%. The screening result for spiked sample shows all the spiked tranquilizers could be correctly identified with low deviations of retention time (< 0.1 min) and mass (< 3 mDa) and high degrees of isotope pattern match (> 89.5%). The developed method was further applied for the analysis of 50 real dairy products, and no positive sample was detected.Twenty tranquilizers were separated within 9 min using UPLC/HRTOF-MS. The deviations of Rt and MW were less than 0.1 min and 3 mDa. The LODs and LOQs were 0.3–1.5 ng mL−1 and 1–5 ng mL−1. The linear ranges were 1–500 ng mL−1 or 5–1000 ng mL−1.
Co-reporter:Si-Yuan PENG, Jie ZHANG, Mei-Ping TIAN, Zhan-Lin WANG, He-Qing SHEN
Chinese Journal of Analytical Chemistry 2012 Volume 40(Issue 8) pp:1201-1206
Publication Date(Web):August 2012
DOI:10.1016/S1872-2040(11)60566-5
DNA methylation is one of the most common epigenetic modifications. Global genomic DNA hypomethylation is considered to induce chromosome instability and high incidence of genetic mutation, which is closely related to the emergence and development of cancer. In this study, a method based on liquid chromatography-electrospray ionization tandem mass spectrometry (LC-ESI-MS/MS) was developed to determine global DNA methylation ratios in various biological samples. DNA was extracted from the samples and hydrolyzed by three specific enzymes into single nucleosides. Liquid chromatography coupled to tandem mass spectrometry was used to measure the concentrations of 2'-deoxycytidine and 2'-deoxy-5-methylcytidine respectively, so as to calculate the global DNA methylation ratios. The developed method was further used to explore the global DNA methylation ratios in normal human liver cell L-02 exposed to perfluorooctane sulfonate (PFOS) and plasma samples of 10 hepatocellular carcinoma patients and 10 healthy participants as controls. This approach has high sensitivity and stability, and is easy to be operated, enabling us to analyze the global DNA methylation ratios in various biological samples, especially those valuable samples (such as serum, plasma et al.) with extremely low concentration of DNA.
Co-reporter:Xiaofei Wang, Shoufang Jiang, Ying Liu, Xiaoyan Du, Weibing Zhang, Jie Zhang, Heqing Shen
Science of The Total Environment (15 August 2017) Volume 592() pp:41-50
Publication Date(Web):15 August 2017
DOI:10.1016/j.scitotenv.2017.03.064
•PM2.5 induced the significant and comprehensive alteration of pulmonary metabolome.•Fifty potential metabolic biomarkers, mainly lipids and nucleotides, were identified from aqueous and organic extracts.•PM2.5 applied pulmonary toxicity through disturbing pro-oxidant/antioxidant balance.Airborne fine particulate matter (PM2.5) has been closely related with a variety of lung diseases. Although some modes of action (e.g. oxidative stress, inflammations) have been proposed, but the pulmonary toxicological mechanism remains obscure. In this paper, in order to understand the comprehensive pulmonary response to PM2.5 stress, a non-targeted high-throughput metabolomics strategy was adopted to characterize the overall metabolic changes and relevant toxicological pathways. PM2.5 samples were collected from Tangshan, one of the most polluted cities in China. Adult male rats were treated with PM2.5 suspension once a week at the dose of 1 mg/kg/week through intratracheal instillation in three months. Aqueous and organic metabolite extracts of the lung tissues were subjected to metabolomics analysis using ultra-high performance liquid chromatograph/mass spectrometry. Along with a significant increase of oxidative stress, significant metabolome alterations were observed in the lung tissues of the treated rats. Nineteen metabolites were found decreased and 31 metabolites increased, which are mainly involved in lipid and nucleotide metabolism. Integrated pathway analysis suggests that PM2.5 can induce pulmonary toxicity through disturbing pro-oxidant/antioxidant balance, which may further correlate with metabolism changes of phospholipid, glycerophospholipid, sphingolipid and purine. These findings improve our understanding of the toxicological pathways of PM2.5 exposure.Download high-res image (225KB)Download full-size image

![1-?Propanaminium, 3-?carboxy-?2-?[(3-?hydroxy-?1-?oxohexadecyl)?oxy]?-?N,?N,?N-?trimethyl-?, inner salt, (2R)?-](http://img.cochemist.com/ccimg/195300/195207-76-2.png)
![1-?Propanaminium, 3-?carboxy-?2-?[(3-?hydroxy-?1-?oxohexadecyl)?oxy]?-?N,?N,?N-?trimethyl-?, inner salt, (2R)?-](http://img.cochemist.com/ccimg/195300/195207-76-2_b.png)
![Benzo[10,11]chryseno[3,4-b]oxirene-7,8-diol,7,8,8a,9a-tetrahydro-, (7R,8S,8aS,9aR)-rel-](http://img.cochemist.com/ccimg/59000/58917-67-2.png)
![Benzo[10,11]chryseno[3,4-b]oxirene-7,8-diol,7,8,8a,9a-tetrahydro-, (7R,8S,8aS,9aR)-rel-](http://img.cochemist.com/ccimg/59000/58917-67-2_b.png)


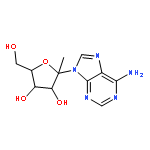




![3-[[(1S)-1,3-dicarboxypropyl]carbamoyl]-3-hydroxy-pentanedioic acid](http://img.cochemist.com/ccimg/73600/73590-26-8.png)
![3-[[(1S)-1,3-dicarboxypropyl]carbamoyl]-3-hydroxy-pentanedioic acid](http://img.cochemist.com/ccimg/73600/73590-26-8_b.png)
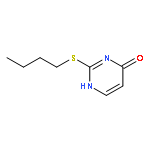
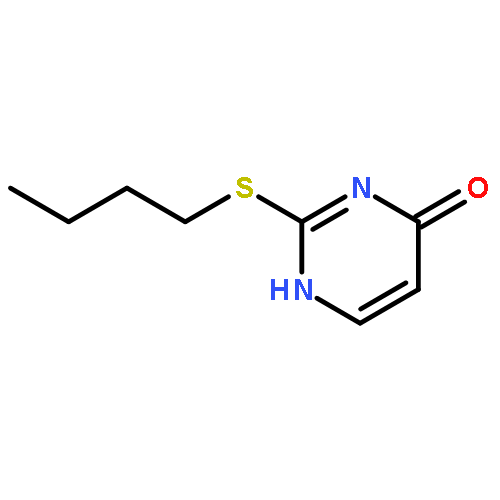
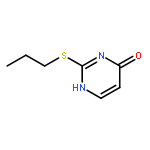
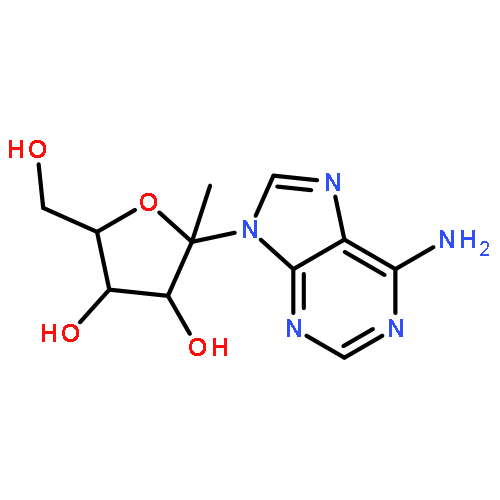
![Acetamide,N-[3-[2-(formylamino)-5-methoxyphenyl]-3-oxopropyl]-](http://img.cochemist.com/ccimg/52500/52450-38-1.png)
![Acetamide,N-[3-[2-(formylamino)-5-methoxyphenyl]-3-oxopropyl]-](http://img.cochemist.com/ccimg/52500/52450-38-1_b.png)





![Ferrate(4-), [7,12-bis[(1S)-1-[(2-amino-2-carboxyethyl)thio]ethyl]-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(6-)-κN21,κN22,κN23,κN24]-,](/data/chemimg/488700/26598-29-8.png)
![Ferrate(4-), [7,12-bis[(1S)-1-[(2-amino-2-carboxyethyl)thio]ethyl]-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(6-)-κN21,κN22,κN23,κN24]-,](/data/chemimg/488700/26598-29-8_b.png)
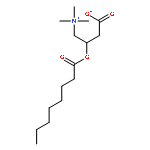

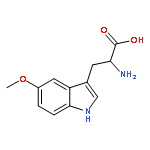

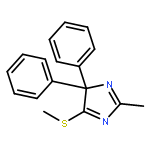
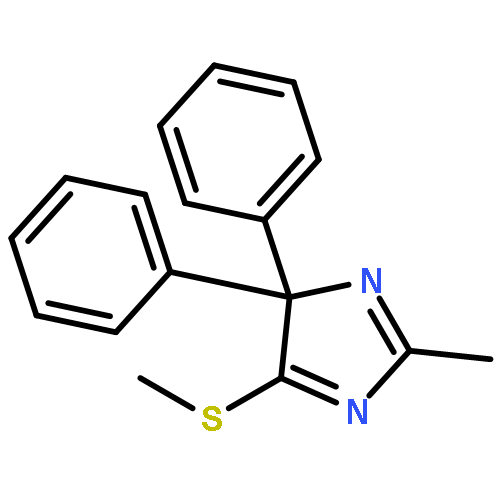
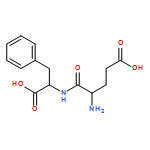
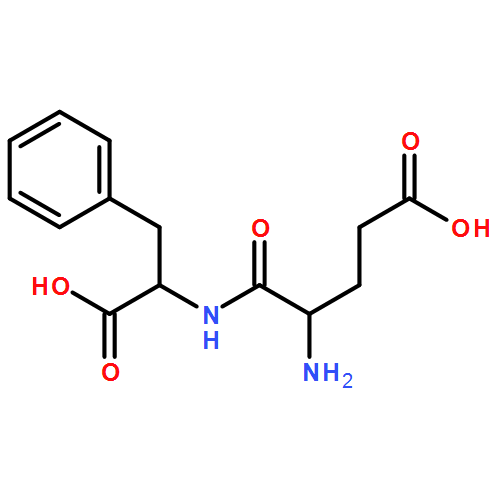


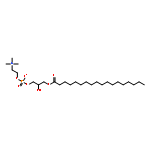




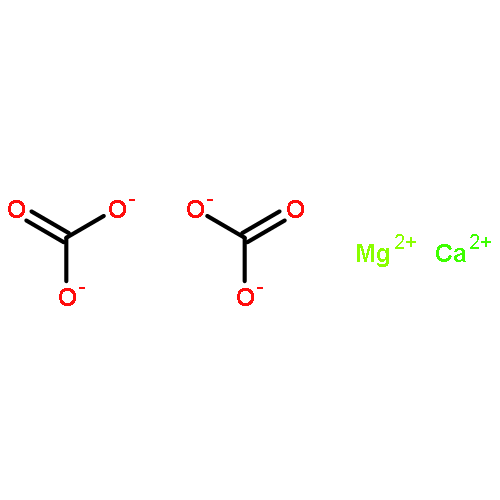




![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8_b.png)


![7-Methylbenzo[b]thiophene](http://img.cochemist.com/ccimg/14400/14315-15-2.png)
![7-Methylbenzo[b]thiophene](http://img.cochemist.com/ccimg/14400/14315-15-2_b.png)




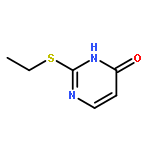
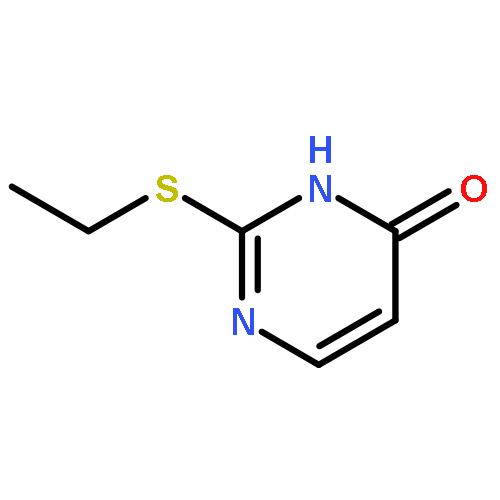

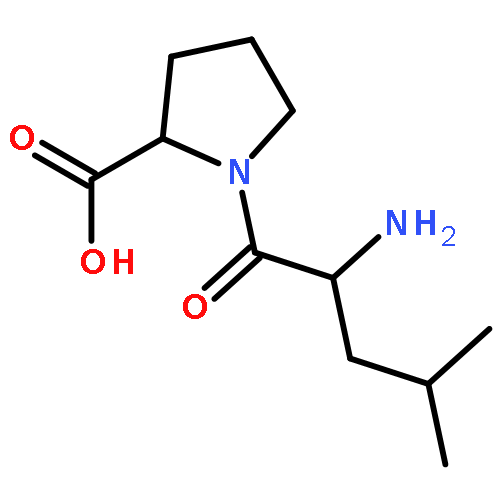
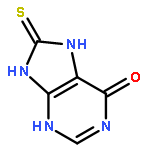
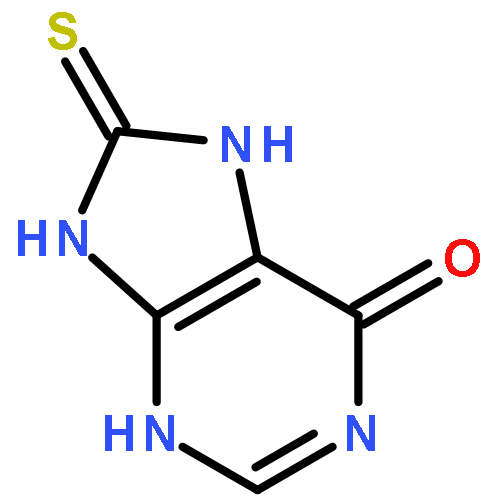


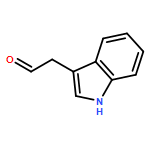
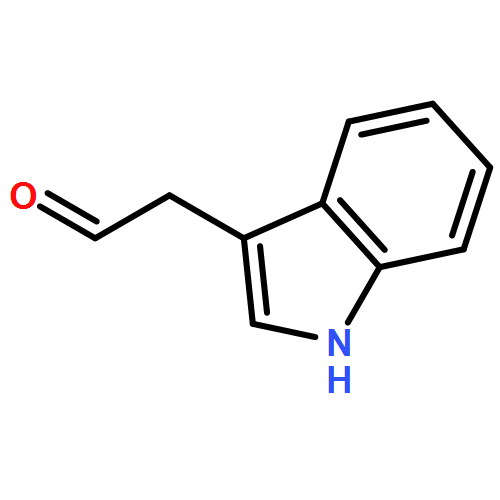
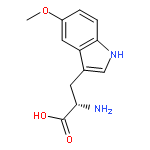
![L-Aspartic acid,N-[[[(4S)-4-amino-4-carboxybutyl]amino]iminomethyl]-](http://img.cochemist.com/ccimg/2400/2387-71-5.png)
![L-Aspartic acid,N-[[[(4S)-4-amino-4-carboxybutyl]amino]iminomethyl]-](http://img.cochemist.com/ccimg/2400/2387-71-5_b.png)
![1-Propanaminium,3-carboxy-N,N,N-trimethyl-2-[(1-oxohexadecyl)oxy]-, inner salt, (2R)-](http://img.cochemist.com/ccimg/2400/2364-67-2.png)
![1-Propanaminium,3-carboxy-N,N,N-trimethyl-2-[(1-oxohexadecyl)oxy]-, inner salt, (2R)-](http://img.cochemist.com/ccimg/2400/2364-67-2_b.png)



![Ethanesulfonic acid,2-[[(3a,5b,12a)-3,12-dihydroxy-24-oxocholan-24-yl]amino]-](http://img.cochemist.com/ccimg/600/516-50-7.png)
![Ethanesulfonic acid,2-[[(3a,5b,12a)-3,12-dihydroxy-24-oxocholan-24-yl]amino]-](http://img.cochemist.com/ccimg/600/516-50-7_b.png)



![Naphtho[2,3-b]thiophene](http://img.cochemist.com/ccimg/300/268-77-9.png)
![Naphtho[2,3-b]thiophene](http://img.cochemist.com/ccimg/300/268-77-9_b.png)
![Benzo[b]naphtho[2,1-d]thiophene](http://img.cochemist.com/ccimg/300/239-35-0.png)
![Benzo[b]naphtho[2,1-d]thiophene](http://img.cochemist.com/ccimg/300/239-35-0_b.png)