Co-reporter:M. Daniel Ricketts, Ronen Marmorstein
Journal of Molecular Biology 2017 Volume 429, Issue 13(Volume 429, Issue 13) pp:
Publication Date(Web):30 June 2017
DOI:10.1016/j.jmb.2016.11.010
•Molecular basis for the assembly of the H3.3-specific HIRA histone chaperone complex•Structural analysis of the mechanism underlying H3.3-specific binding by the HIRA complex•Proposed model for (H3.3/H4)2 tetramer formation by the HIRA complexIncorporation of variant histone sequences, in addition to post-translational modification of histones, serves to modulate the chromatin environment. Different histone chaperone proteins mediate the storage and chromatin deposition of variant histones. Although the two non-centromeric histone H3 variants, H3.1 and H3.3, differ by only 5 aa, replacement of histone H3.1 with H3.3 can modulate the transcription for highly expressed and developmentally required genes, lead to the formation of repressive heterochromatin, or aid in DNA and chromatin repair. The human histone cell cycle regulator (HIRA) complex composed of HIRA, ubinuclein-1, CABIN1, and transiently anti-silencing function 1, forms one of the two complexes that bind and deposit H3.3/H4 into chromatin. A number of recent biochemical and structural studies have revealed important details underlying how these proteins assemble and function together as a multiprotein H3.3-specific histone chaperone complex. Here, we present a review of existing data and present a new model for the assembly of the HIRA complex and for the HIRA-mediated incorporation of H3.3/H4 into chromatin.Download high-res image (116KB)Download full-size image
Co-reporter:Cheryl E. McCullough and Ronen Marmorstein
ACS Chemical Biology 2016 Volume 11(Issue 3) pp:632
Publication Date(Web):November 11, 2015
DOI:10.1021/acschembio.5b00841
Acetylation is a post-translational modification (PTM) that regulates chromatin dynamics and function. Dysregulation of acetylation or acetyltransferase activity has been correlated with several human diseases. Many, if not all, histone acetyltransferases (HATs) are regulated in part through tethered domains, association with binding partners, or post-translational modification, including predominantly acetylation. This review focuses on what is currently understood at the molecular level of HAT regulation as it occurs via binding partners, associated domains, and autoacetylation.
Co-reporter:Michael Grasso, Michelle A. Estrada, Christian Ventocilla, Minu Samanta, Jasna Maksimoska, Jessie Villanueva, Jeffrey D. Winkler, and Ronen Marmorstein
ACS Chemical Biology 2016 Volume 11(Issue 10) pp:2876
Publication Date(Web):August 29, 2016
DOI:10.1021/acschembio.6b00529
The BRAF kinase, within the mitogen activated protein kinase (MAPK) signaling pathway, harbors activating mutations in about half of melanomas and to a significant extent in many other cancers. A single valine to glutamic acid substitution at residue 600 (BRAFV600E) accounts for about 90% of these activating mutations. While BRAFV600E-selective small molecule inhibitors, such as debrafenib and vemurafenib, have shown therapeutic benefit, almost all patients develop resistance. Resistance often arises through reactivation of the MAPK pathway, typically through mutation of upstream RAS, downstream MEK, or splicing variants. RAF kinases signal as homo- and heterodimers, and another complication associated with small molecule BRAFV600E inhibition is drug-induced allosteric activation of a wild-type RAF subunit (BRAF or CRAF) of the kinase dimer, a process called “transactivation” or “paradoxical activation.” Here, we used BRAFV600E and vemurafenib as a model system to develop chemically linked kinase inhibitors to lock RAF dimers in an inactive conformation that cannot undergo transactivation. This structure-based design effort resulted in the development of Vem-BisAmide-2, a compound containing two vemurafenib molecules connected by a bis amide linker. We show that Vem-BisAmide-2 has comparable inhibitory potency as vemurafenib to BRAFV600E both in vitro and in cells but promotes an inactive dimeric BRAFV600E conformation unable to undergo transactivation. The crystal structure of a BRAFV600E/Vem-BisAmide-2 complex and associated biochemical studies reveal the molecular basis for how Vem-BisAmide-2 mediates selectivity for an inactive over an active dimeric BRAFV600E conformation. These studies have implications for targeting BRAFV600E/RAF heterodimers and other kinase dimers for therapy.
Co-reporter:Jie Qin; Rajathees Rajaratnam; Li Feng; Jemilat Salami; Julie S. Barber-Rotenberg; John Domsic; Patricia Reyes-Uribe; Haiying Liu; Weiwei Dang; Shelley L. Berger; Jessie Villanueva; Eric Meggers
Journal of Medicinal Chemistry 2015 Volume 58(Issue 1) pp:305-314
Publication Date(Web):October 30, 2014
DOI:10.1021/jm5011868
Aberrant activation of S6 kinase 1 (S6K1) is found in many diseases, including diabetes, aging, and cancer. We developed ATP competitive organometallic kinase inhibitors, EM5 and FL772, which are inspired by the structure of the pan-kinase inhibitor staurosporine, to specifically inhibit S6K1 using a strategy previously used to target other kinases. Biochemical data demonstrate that EM5 and FL772 inhibit the kinase with IC50 value in the low nanomolar range at 100 μM ATP and that the more potent FL772 compound has a greater than 100-fold specificity over S6K2. The crystal structures of S6K1 bound to staurosporine, EM5, and FL772 reveal that the EM5 and FL772 inhibitors bind in the ATP binding pocket and make S6K1-specific contacts, resulting in changes to the p-loop, αC helix, and αD helix when compared to the staurosporine-bound structure. Cellular data reveal that FL772 is able to inhibit S6K phosphorylation in yeast cells. Together, these studies demonstrate that potent, selective, and cell permeable S6K1 inhibitors can be prepared and provide a scaffold for future development of S6K inhibitors with possible therapeutic applications.
Co-reporter:Adam S. Olia, Kristi Barker, Cheryl E. McCullough, Hsin-Yao Tang, David W. Speicher, Ji Qiu, Joshua LaBaer, and Ronen Marmorstein
ACS Chemical Biology 2015 Volume 10(Issue 9) pp:2034
Publication Date(Web):June 17, 2015
DOI:10.1021/acschembio.5b00342
Acetylation is a post-translational modification that occurs on thousands of proteins located in many cellular organelles. This process mediates many protein functions and modulates diverse biological processes. In mammalian cells, where acetyl-CoA is the primary acetyl donor, acetylation in the mitochondria is thought to occur by chemical means due to the relatively high concentration of acetyl-CoA located in this organelle. In contrast, acetylation outside of the mitochondria is thought to be mediated predominantly by acetyltransferase enzymes. Here, we address the possibility that nonenzymatic chemical acetylation outside of the mitochondria may be more common than previously appreciated. We employed the Nucleic Acid Programmable Protein Array platform to perform an unbiased screen for human proteins that undergo chemical acetylation, which resulted in the identification of a multitude of proteins with diverse functions and cellular localization. Mass spectrometry analysis revealed that basic residues typically precede the acetylated lysine in the −7 to −3 position, and we show by mutagenesis that these basic residues contribute to chemical acetylation capacity. We propose that these basic residues lower the pKa of the substrate lysine for efficient chemical acetylation. Many of the identified proteins reside outside of the mitochondria and have been previously demonstrated to be acetylated in vivo. As such, our studies demonstrate that chemical acetylation occurs more broadly throughout the eukaryotic cell than previously appreciated and suggests that this post-translational protein modification may have more diverse roles in protein function and pathway regulation.
Co-reporter:Kimberly A. Malecka, Daniela Fera, David C. Schultz, Santosh Hodawadekar, Melvin Reichman, Preston S. Donover, Maureen E. Murphy, and Ronen Marmorstein
ACS Chemical Biology 2014 Volume 9(Issue 7) pp:1603
Publication Date(Web):May 22, 2014
DOI:10.1021/cb500229d
Cervical cancer is the sixth most common cancer in women worldwide and the leading cause of women’s death in developing countries. Nearly all cervical cancers are associated with infection of the human papillomavirus (HPV). This sexually transmitted pathogen disrupts the cell cycle via two oncoproteins: E6 and E7. Cells respond to E7-mediated degradation of pRB by upregulating the p53 tumor suppressor pathway. However, E6 thwarts this response by binding to the cellular E6-Associating Protein (E6AP) and targeting p53 for degradation. These two virus-facilitated processes pave the way for cellular transformation. Prophylactic HPV vaccines are available, but individuals already infected with HPV lack drug-based therapeutic options. To fill this void, we sought to identify small molecule inhibitors of the E6–E6AP interaction. We designed an ELISA-based high throughput assay to rapidly screen compound libraries, and hits were confirmed in several orthogonal biochemical and cell-based assays. Over 88,000 compounds were screened; 30 had in vitro potencies in the mid-nanomolar to mid-micromolar range and were classified as validated hits. Seven of these hits inhibited p53 degradation in cell lines with HPV-integrated genomes. Two compounds of similar scaffold successfully blocked p53 degradation and inhibited cell proliferation in cells stably transfected with E6. Together, these studies suggest that small molecules can successfully block E6-dependent p53 degradation and restore p53 activity. The compounds identified here constitute attractive starting points for further medicinal chemistry efforts and development into beneficial therapeutics.
Co-reporter:Jasna Maksimoska, Dario Segura-Peña, Philip A. Cole, and Ronen Marmorstein
Biochemistry 2014 Volume 53(Issue 21) pp:
Publication Date(Web):May 12, 2014
DOI:10.1021/bi500380f
The p300 and CBP transcriptional coactivator paralogs (p300/CBP) regulate a variety of different cellular pathways, in part, by acetylating histones and more than 70 non-histone protein substrates. Mutation, chromosomal translocation, or other aberrant activities of p300/CBP are linked to many different diseases, including cancer. Because of its pleiotropic biological roles and connection to disease, it is important to understand the mechanism of acetyl transfer by p300/CBP, in part so that inhibitors can be more rationally developed. Toward this goal, a structure of p300 bound to a Lys-CoA bisubstrate HAT inhibitor has been previously elucidated, and the enzyme’s catalytic mechanism has been investigated. Nonetheless, many questions underlying p300/CBP structure and mechanism remain. Here, we report a structural characterization of different reaction states in the p300 activity cycle. We present the structures of p300 in complex with an acetyl-CoA substrate, a CoA product, and an acetonyl-CoA inhibitor. A comparison of these structures with the previously reported p300/Lys-CoA complex demonstrates that the conformation of the enzyme active site depends on the interaction of the enzyme with the cofactor, and is not apparently influenced by protein substrate lysine binding. The p300/CoA crystals also contain two poly(ethylene glycol) moieties bound proximal to the cofactor binding site, implicating the path of protein substrate association. The structure of the p300/acetonyl-CoA complex explains the inhibitory and tight binding properties of the acetonyl-CoA toward p300. Together, these studies provide new insights into the molecular basis of acetylation by p300 and have implications for the rational development of new small molecule p300 inhibitors.
Co-reporter:Svein Isungset Støve, Robert S. Magin, Håvard Foyn, Bengt Erik Haug, ... Thomas Arnesen
Structure (6 July 2016) Volume 24(Issue 7) pp:1044-1056
Publication Date(Web):6 July 2016
DOI:10.1016/j.str.2016.04.020
•Crystal structure of Naa60, a unique membrane-associated N-terminal acetyltransferase•Naa60-specific loops are important for enzyme stability and substrate binding•Naa60 β6-β7 loop mediates dimer to monomer transition for substrate-specific binding•A novel CoA-peptide bisubstrate Naa60-inhibitor was developed and co-crystallizedN-Terminal acetylation is a common and important protein modification catalyzed by N-terminal acetyltransferases (NATs). Six human NATs (NatA–NatF) contain one catalytic subunit each, Naa10 to Naa60, respectively. In contrast to the ribosome-associated NatA to NatE, NatF/Naa60 specifically associates with Golgi membranes and acetylates transmembrane proteins. To gain insight into the molecular basis for the function of Naa60, we developed an Naa60 bisubstrate CoA-peptide conjugate inhibitor, determined its X-ray structure when bound to CoA and inhibitor, and carried out biochemical experiments. We show that Naa60 adapts an overall fold similar to that of the catalytic subunits of ribosome-associated NATs, but with the addition of two novel elongated loops that play important roles in substrate-specific binding. One of these loops mediates a dimer to monomer transition upon substrate-specific binding. Naa60 employs a catalytic mechanism most similar to Naa50. Collectively, these data reveal the molecular basis for Naa60-specific acetyltransferase activity with implications for its Golgi-specific functions.Download high-res image (293KB)Download full-size image
Co-reporter:Robert S. Magin, Glen P. Liszczak, Ronen Marmorstein
Structure (3 February 2015) Volume 23(Issue 2) pp:332-341
Publication Date(Web):3 February 2015
DOI:10.1016/j.str.2014.10.025
•NatD is among the most selective N-terminal acetyltransferases (NATs)•NatD-specific structural features accommodate its high substrate selectivity•NatD substrate site is tailored for a Ser-Gly-Arg-Gly histone H4/H2A N terminus•Studies have implications for understanding substrate-specific acetylation by NATsN-terminal acetylation is among the most common protein modifications in eukaryotes and is mediated by evolutionarily conserved N-terminal acetyltransferases (NATs). NatD is among the most selective NATs; its only known substrates are histones H4 and H2A, containing the N-terminal sequence SGRGK in humans. Here we characterize the molecular basis for substrate-specific acetylation by NatD by reporting its crystal structure bound to cognate substrates and performing related biochemical studies. A novel N-terminal segment wraps around the catalytic core domain to make stabilizing interactions, and the α1-α2 and β6-β7 loops adopt novel conformations to properly orient the histone N termini in the binding site. Ser1 and Arg3 of the histone make extensive contacts to highly conserved NatD residues in the substrate binding pocket, and flanking glycine residues also appear to contribute to substrate-specific binding by NatD, together defining a Ser-Gly-Arg-Gly recognition sequence. These studies have implications for understanding substrate-specific acetylation by NAT enzymes.Download high-res image (679KB)Download full-size image
.jpg)
![N-(3-(5-Bromo-1-(2,6-dichlorobenzoyl)-1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluorophenyl)propane-1-sulfonamide](http://img.cochemist.com/ccimg/1263000/1262985-24-9.png)
![N-(3-(5-Bromo-1-(2,6-dichlorobenzoyl)-1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluorophenyl)propane-1-sulfonamide](http://img.cochemist.com/ccimg/1263000/1262985-24-9_b.png)
![N-(3-(5-(4-Chlorophenyl)-1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluorophenyl)propane-1-sulfonamide](http://img.cochemist.com/ccimg/918600/918504-65-1.png)
![N-(3-(5-(4-Chlorophenyl)-1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluorophenyl)propane-1-sulfonamide](http://img.cochemist.com/ccimg/918600/918504-65-1_b.png)
![(3AR,4R,5R,6AS)-4-FORMYL-2-OXOHEXAHYDRO-2H-CYCLOPENTA[B]FURAN-5-Y<WBR />L 4-BIPHENYLCARBOXYLATE](http://img.cochemist.com/ccimg/100/72-89-9.png)
![(3AR,4R,5R,6AS)-4-FORMYL-2-OXOHEXAHYDRO-2H-CYCLOPENTA[B]FURAN-5-Y<WBR />L 4-BIPHENYLCARBOXYLATE](http://img.cochemist.com/ccimg/100/72-89-9_b.png)
























































































































































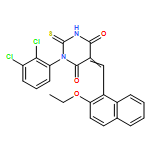



![1-Propanone,3-[(1-methylethyl)(phenylmethyl)amino]-1-(2-naphthalenyl)-](http://img.cochemist.com/ccimg/273800/273727-89-2.png)
![1-Propanone,3-[(1-methylethyl)(phenylmethyl)amino]-1-(2-naphthalenyl)-](http://img.cochemist.com/ccimg/273800/273727-89-2_b.png)








![Benzenepropanenitrile, a-[bis(methylthio)methylene]-b-oxo-3-(trifluoromethyl)-](http://img.cochemist.com/ccimg/116500/116492-97-8.png)
![Benzenepropanenitrile, a-[bis(methylthio)methylene]-b-oxo-3-(trifluoromethyl)-](http://img.cochemist.com/ccimg/116500/116492-97-8_b.png)


![2-Phenylbenzo[d][1,2]selenazol-3(2H)-one](http://img.cochemist.com/ccimg/61000/60940-34-3.png)
![2-Phenylbenzo[d][1,2]selenazol-3(2H)-one](http://img.cochemist.com/ccimg/61000/60940-34-3_b.png)
![Imidazo[2,1-b]naphtho[2,3-d]thiazole-5,10-dione](http://img.cochemist.com/ccimg/59800/59774-66-2.png)
![Imidazo[2,1-b]naphtho[2,3-d]thiazole-5,10-dione](http://img.cochemist.com/ccimg/59800/59774-66-2_b.png)




![[1,1'-Biphenyl]-4-sulfonamide](http://img.cochemist.com/ccimg/4400/4371-23-7.png)
![[1,1'-Biphenyl]-4-sulfonamide](http://img.cochemist.com/ccimg/4400/4371-23-7_b.png)



