Co-reporter:Keith Izod, Corinne Wills, Emma Anderson, Ross W. Harrington, and Michael R. Probert
Organometallics 2014 Volume 33(Issue 19) pp:5283-5294
Publication Date(Web):September 5, 2014
DOI:10.1021/om5005995
The reaction between iPr2PCl and Ph2P(BH3)CH2Li gives the mixed phosphine/phosphine-borane Ph2P(BH3)CH2PiPr2 (1a) in good yield. Thermolysis of 1a leads to borane migration and the formation of Ph2PCH2P(BH3)iPr2 (2a) along with small amounts of Ph2P(BH3)CH2P(BH3)iPr2 (3a) and Ph2PCH2PiPr2 (4a). Compound 3a may be synthesized directly from the reaction of 1a with BH3·SMe2, while 4a can be prepared cleanly by heating 1a in methanol under reflux. Kinetic studies on the conversion of 1a to 2a reveal the reaction to be apparently first order in 1a, suggesting a dissociative process, and yield the activation parameters ΔH⧧ = 63 ± 8 kJ mol–1, ΔS⧧ = −145 ± 24 J K–1 mol–1, and ΔG⧧ = 106 ± 8 kJ mol–1, the negative entropy of activation conversely suggesting an associative process. DFT studies suggest that concerted migration of borane within a molecule of 1a is disfavored, but that both the dissociative and associative mechanisms for borane migration operate simultaneously. Metalation of 1a–4a with nBuLi in the presence of tmeda gives the complexes [{Ph2P(BH3)}CHPiPr2]Li(tmeda) (1b), [Ph2PCH{P(BH3)iPr2}]Li(tmeda) (2b), [{Ph2P(BH3)}CH{P(BH3)iPr2}]Li(tmeda) (3b), and [Ph2PCHPiPr2]Li(tmeda) (4b), respectively, which adopt similar structures in the solid state. Analysis of the crystal structures suggests that the phosphine-borane groups stabilize the adjacent charge to a greater extent than the phosphine groups. This is supported by DFT calculations, which show that the greatest delocalization of negative charge from the carbanion is into the P–C(Ph) or P–C(Pr) σ*-orbitals of the phosphine-borane substituents.
Co-reporter:Keith Izod, Corinne Wills, Ross W. Harrington, William Clegg
Journal of Organometallic Chemistry 2013 725() pp: 11-14
Publication Date(Web):
DOI:10.1016/j.jorganchem.2012.12.010



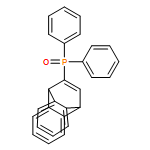
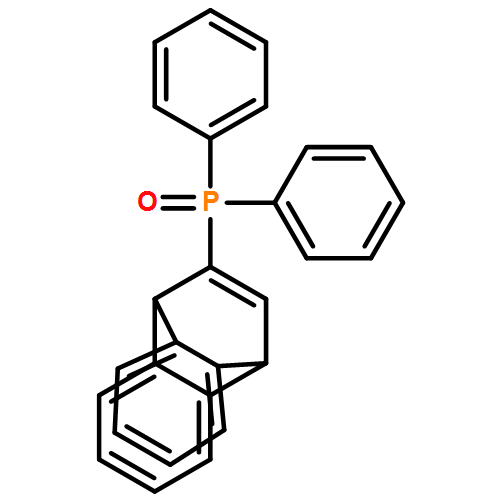




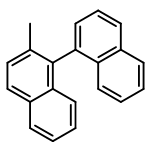











![PHOSPHONOUS DICHLORIDE, [BIS(TRIMETHYLSILYL)METHYL]-](http://img.cochemist.com/ccimg/76600/76505-20-9.png)
![PHOSPHONOUS DICHLORIDE, [BIS(TRIMETHYLSILYL)METHYL]-](http://img.cochemist.com/ccimg/76600/76505-20-9_b.png)








![Isobenzofuran, 1,3-dihydro-1-[(4-methylphenyl)methylene]-](http://img.cochemist.com/ccimg/64500/64421-13-2.png)
![Isobenzofuran, 1,3-dihydro-1-[(4-methylphenyl)methylene]-](http://img.cochemist.com/ccimg/64500/64421-13-2_b.png)


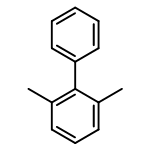






![Phosphine, [[bis(1-methylethyl)phosphino]methyl]diphenyl-](http://img.cochemist.com/ccimg/62300/62263-67-6.png)
![Phosphine, [[bis(1-methylethyl)phosphino]methyl]diphenyl-](http://img.cochemist.com/ccimg/62300/62263-67-6_b.png)


























![Ethanone,1-[1,1'-biphenyl]-2-yl-](http://img.cochemist.com/ccimg/2200/2142-66-7.png)
![Ethanone,1-[1,1'-biphenyl]-2-yl-](http://img.cochemist.com/ccimg/2200/2142-66-7_b.png)