Co-reporter:Chun-lai Ren;Roland Hager;Robert Schlapak;Stefan Howorka
Langmuir October 27, 2015 Volume 31(Issue 42) pp:11491-11501
Publication Date(Web):2017-2-22
DOI:10.1021/acs.langmuir.5b02674
Poly(ethylene glycol) (PEG) nanofilms are used to avert the nonspecific binding of biomolecules on substrate surfaces in biomedicine and bioanalysis including modern fluorescence-based DNA sensing and sequencing chips. A fundamental and coherent understanding of the interactions between fluorophore-tagged DNA, PEG-films, and substrates in terms of molecular and energetic factors is, however, missing. Here we explore a large parameter space to elucidate how PEG layers passivate metal oxide surfaces against Cy3-labeled DNA probes. The driving force for probe adsorption is found to be the affinity of the fluorophore to the substrate, while the high-quality PEG films prevent adsorption to bare ITO surfaces. The amount of nonrepelled, surface-bound DNA strongly depends on oligonucleotide size, PEG chain length, and incubation temperature. To explain these observations, we develop an experimentally validated theory to provide a microscopic picture of the PEG layer and show that adsorbed DNA molecules reside within the film by end-tethering the fluorophore to the ITO surface. To compensate for the local accumulation of negatively charged DNA, counterions condense on the adsorbed probes within the layer. The model furthermore explains that surface passivation is governed by the interdependence of molecular size, conformation, charge, ion condensation, and environmental conditions. We finally report for the first time on the detailed thermodynamic values that show how adsorption results from a balance between large opposing energetic factors. The insight of our study can be applied to rationally engineer PEG nanolayers for improved functional performance in DNA analysis schemes and may be expanded to other polymeric thin films.
Co-reporter:Kai Huang and Igal Szleifer
Journal of the American Chemical Society May 10, 2017 Volume 139(Issue 18) pp:6422-6422
Publication Date(Web):April 19, 2017
DOI:10.1021/jacs.7b02057
Nature uses the interplay between hydrophobic and electrostatic interactions of disordered proteins to orchestrate complicated molecular gates such as the nuclear pore complex to control the transport of biological masses. Inspired by nature, we here theoretically show that well-defined gate shape, sensitive response to pH and salt concentration, and selectivity in cargo transport can be simultaneously achieved by grafting amphiphilic diblock copolymers made of sequence-controlled hydrophobic and ionizable monomers on the inner surface of solid-state nanopore. As a result, multiple functions such as ionic gating and molecular filtering can be implemented into one single copolymer nanogate. The gate structure and thermodynamics is a result of the self-assembly of the sequence-designed copolymer in the confined geometry that minimizes the free energy of the system. Our theory further predicts a phase transition and discontinuous charge regulation of the confined copolymer that allows logical gating in biosensors and nanofluidic devices. As an example of application, a nanolocker with the potential of molecular pumping has also been designed with the cooperation of two amphiphilic copolymer gates. Our results highlight the importance of polymer sequence in nanogating, and these insights can be used to guide the rational design of polymer-coated smart nanopores.
Co-reporter:Gabriel S. Longo, Monica Olvera de la Cruz, and Igal Szleifer
Langmuir December 23, 2014 Volume 30(Issue 50) pp:15335-15344
Publication Date(Web):December 1, 2014
DOI:10.1021/la5040382
We present a molecular theory to study the adsorption of different species within pH-sensitive hydrogel nanofilms. The theoretical framework allows for a molecular-level description of all the components of the system, and it explicitly accounts for the acid–base equilibrium. We concentrate on the adsorption of hexahistidine, one of the most widely used tags in bio-related systems, particularly in chromatography of proteins. The adsorption of hexahistidine within a grafted polyacid hydrogel film shows a nonmonotonic dependence on the solution pH. Depending on the salt concentration, the density of the polymer network, and the bulk concentration of peptide, substantial adsorption is predicted in the intermediate pH range where both the network and the amino acids are charged. To enhance the electrostatic attractions, the acid–base equilibrium of adsorbed hexahistidine is shifted significantly, increasing the degree of charge of the residues as compared to the bulk solution. Such a shift depends critically on the conditions of the environment at the nanoscale. At the same time, the degree of dissociation of the network becomes that of the isolated acid group in a dilute solution, which means that the network is considerably more charged than when there is no adsorbate molecules. This work provides fundamental information on the physical chemistry behind the adsorption behavior and the response of the hydrogel film. This information can be useful in designing new materials for the purification or separation/immobilization of histidine-tagged proteins.
Co-reporter:Simona Morochnik;Rikkert J. Nap;Guillermo A. Ameer
Soft Matter (2005-Present) 2017 vol. 13(Issue 37) pp:6322-6331
Publication Date(Web):2017/09/27
DOI:10.1039/C7SM01538K
Herein, we develop a molecular theory to examine a class of pH and temperature-responsive tethered polymer layers. The response of pH depends on intramolecular charge repulsion of weakly acidic monomers and the response of temperature depends on hydrogen bonding between polymer monomers and water molecules akin to the behavior of water-soluble polymers such as PEG (poly-ethylene glycol) or NIPAAm (n-isopropylacrylamide). We investigate the changes in structural behavior that result for various end-tethered copolymers: pH/T responsive monomers alone, in alternating sequence with hydrophobic monomers, and as 50/50 diblocks with hydrophobic monomers. We find that the sequence and location of hydrophobic units play a critical role in the thermodynamic stability and structural behavior of these responsive polymer layers. Additionally, the polymers exhibit tunable collapse when varying the surface coverage, location and sequence of hydrophobic units as a function of temperature and pH. As far as we know, our results present the first molecularly detailed theory for end-tethered polymers that are both pH and temperature-responsive via hydrogen bonding. We propose that this work holds predictive power for the guided design of future biomaterials.
Co-reporter:Mario Tagliazucchi, Igal Szleifer
Materials Today 2015 Volume 18(Issue 3) pp:131-142
Publication Date(Web):April 2015
DOI:10.1016/j.mattod.2014.10.020
The last few years have witnessed major advancements in the synthesis, modification, characterization and modeling of nanometer-size solid-state channels and pores. Future applications in sensing, energy conversion and purification technologies will critically rely on qualitative improvements in the control over the selectivity, directionality and responsiveness of these nanochannels and nanopores. It is not surprising, therefore, that researchers in the field seek inspiration in biological ion channels and ion pumps, paradigmatic examples of transport selectivity. This work reviews our current fundamental understanding of the mechanisms of transport of ions and larger cargoes through nanopores and nanochannels by examining recent experimental and theoretical work. It is argued that that structure and transport in biological channels and polyelectrolyte-modified synthetic nanopores are strongly coupled: the structure dictates transport and transport affects the structure. We compare synthetic and biological systems throughout this review to conclude that while they present interesting similarities, they also have striking differences.
Co-reporter:Mario Tagliazucchi
Journal of the American Chemical Society 2015 Volume 137(Issue 39) pp:12539-12551
Publication Date(Web):September 14, 2015
DOI:10.1021/jacs.5b05032
We present systematic studies for the binding of small model proteins to ligands attached to the inner walls of long nanochannels and short nanopores by polymeric tethers. Binding of proteins to specific ligands inside nanometric channels and pores leads to changes in their ionic conductance, which have been exploited in sensors that quantify the concentration of the proteins in solution. The theoretical predictions presented in this work are aimed to provide a fundamental understanding of protein binding under geometrically confined environments and to guide the design of this kind of nanochannel-based sensors. The theory predicts that the fraction of the channel volume filled by bound proteins is a nonmonotonic function of the channel radius, the length of the tethers, the surface density of the ligands and the size of the proteins. Notably, increasing the density of ligands, decreasing the size of the channel or increasing the size of the protein may lead to a decrease of the fraction of the channel volume filled by bound proteins. These results are explained from the incomplete binding of proteins to the ligands due to repulsive protein–protein and protein–ligand steric interactions. Our work suggests strategies to optimize the change in conductance due to protein binding, for example: (i) proteins much smaller than the radius of the channel may effectively block the channel if tethers of appropriate length are used, and (ii) a large decrease in conductance upon protein binding can be achieved if the channel and the protein are oppositely charged.
Co-reporter:Claudio F. Narambuena, Gabriel S. Longo and Igal Szleifer
Soft Matter 2015 vol. 11(Issue 33) pp:6669-6679
Publication Date(Web):13 Jul 2015
DOI:10.1039/C5SM00980D
We develop and apply a molecular theory to study the adsorption of lysozyme on weak polyacid hydrogel films. The theory explicitly accounts for the conformation of the network, the structure of the proteins, the size and shape of all the molecular species, their interactions as well as the chemical equilibrium of each titratable unit of both the protein and the polymer network. The driving forces for adsorption are the electrostatic attractions between the negatively charged network and the positively charged protein. The adsorption is a non-monotonic function of the solution pH, with a maximum in the region between pH 8 and 9 depending on the salt concentration of the solution. The non-monotonic adsorption is the result of increasing negative charge of the network with pH, while the positive charge of the protein decreases. At low pH the network is roughly electroneutral, while at sufficiently high pH the protein is negatively charged. Upon adsorption, the acid–base equilibrium of the different amino acids of the protein shifts in a nontrivial fashion that depends critically on the particular kind of residue and solution composition. Thus, the proteins regulate their charge and enhance adsorption under a wide range of conditions. In particular, adsorption is predicted above the protein isoelectric point where both the solution lysozyme and the polymer network are negatively charged. This behavior occurs because the pH in the interior of the gel is significantly lower than that in the bulk solution and it is also regulated by the adsorption of the protein in order to optimize protein–gel interactions. Under high pH conditions we predict that the protein changes its charge from negative in the solution to positive within the gel. The change occurs within a few nanometers at the interface of the hydrogel film. Our predictions show the non-trivial interplay between acid–base equilibrium, physical interactions and molecular organization under nanoconfined conditions, which leads to non-trivial adsorption behavior that is qualitatively different from what would be predicted from the state of the proteins in the bulk solution.
Co-reporter:Mario Tagliazucchi
The Journal of Physical Chemistry Letters 2015 Volume 6(Issue 18) pp:3534-3539
Publication Date(Web):August 25, 2015
DOI:10.1021/acs.jpclett.5b01315
This Letter investigates voltage-gated nanochannels, where both the potential applied to the conductive membrane containing the channel (membrane potential) and the potential difference between the solutions at both sides of the membrane (transmembrane potential) are independently controlled. The predicted conductance characteristics of these fixed-potential channels dramatically differ from those of the widely studied fixed-charge nanochannels, in which the membrane is insulating and has a fixed surface charge density. The difference arises because the transmembrane potential induces an inhomogeneous charge distribution on the surface of fixed-potential nanochannels. This behavior, related to bipolar electrochemistry, has some interesting and unexpected consequences for ion transport. For example, continuously oscillating the transmembrane potential, while holding the membrane potential at the potential for which it has zero charge in equilibrium, creates fluxes of neutral salt (fluxes of anions and cations in the same direction and number) through the channel, which is an interesting phenomenon for desalination applications.
Co-reporter:K. Baler, O. A. Martin, M. A. Carignano, G. A. Ameer, J. A. Vila, and I. Szleifer
The Journal of Physical Chemistry B 2014 Volume 118(Issue 4) pp:921-930
Publication Date(Web):January 6, 2014
DOI:10.1021/jp409936v
A better understanding of protein aggregation is bound to translate into critical advances in several areas, including the treatment of misfolded protein disorders and the development of self-assembling biomaterials for novel commercial applications. Because of its ubiquity and clinical potential, albumin is one of the best-characterized models in protein aggregation research; but its properties in different conditions are not completely understood. Here, we carried out all-atom molecular dynamics simulations of albumin to understand how electrostatics can affect the conformation of a single albumin molecule just prior to self-assembly. We then analyzed the tertiary structure and solvent accessible surface area of albumin after electrostatically triggered partial denaturation. The data obtained from these single protein simulations allowed us to investigate the effect of electrostatic interactions between two proteins. The results of these simulations suggested that hydrophobic attractions and counterion binding may be strong enough to effectively overcome the electrostatic repulsions between the highly charged monomers. This work contributes to our general understanding of protein aggregation mechanisms, the importance of explicit consideration of free ions in protein solutions, provides critical new insights about the equilibrium conformation of albumin in its partially denatured state at low pH, and may spur significant progress in our efforts to develop biocompatible protein hydrogels driven by electrostatic partial denaturation.
Co-reporter:Igal Szleifer;Mario Tagliazucchi;Emily A. Weiss
PNAS 2014 Volume 111 (Issue 27 ) pp:9751-9756
Publication Date(Web):2014-07-08
DOI:10.1073/pnas.1406122111
Dissipative self-assembly is the emergence of order within a system due to the continuous input of energy. This form of nonequilibrium
self-organization allows the creation of structures that are inaccessible in equilibrium self-assembly. However, design strategies
for dissipative self-assembly are limited by a lack of fundamental understanding of the process. This work proposes a novel
route for dissipative self-assembly via the oscillation of interparticle potentials. It is demonstrated that in the limit
of fast potential oscillations the structure of the system is exactly described by an effective potential that is the time
average of the oscillatory potential. This effective potential depends on the shape of the oscillations and can lead to effective
interactions that are physically inaccessible in equilibrium. As a proof of concept, Brownian dynamics simulations were performed
on a binary mixture of particles coated by weak acids and weak bases under externally controlled oscillations of pH. Dissipative
steady-state structures were formed when the period of the pH oscillations was smaller than the diffusional timescale of the
particles, whereas disordered oscillating structures were observed for longer oscillation periods. Some of the dissipative
structures (dimers, fibers, and honeycombs) cannot be obtained in equilibrium (fixed pH) simulations for the same system of
particles. The transition from dissipative self-assembled structures for fast oscillations to disordered oscillating structures
for slow oscillations is characterized by a maximum in the energy dissipated per oscillation cycle. The generality of the
concept is demonstrated in a second system with oscillating particle sizes.
Co-reporter:Mario Tagliazucchi, Xing Li, Monica Olvera de la Cruz, and Igal Szleifer
ACS Nano 2014 Volume 8(Issue 10) pp:9998
Publication Date(Web):September 15, 2014
DOI:10.1021/nn502008x
Layers of end-grafted weak polyelectrolytes in poor solvent self-organize into a rich variety of structures (such as micelles, micelles coexisting with nonaggregated chains, stripes and layers with solvent-filled holes) due to the subtle competition among hydrophobic, electrostatic and steric interactions and the chemical acid–based equilibria of the weak polyelectrolyte. In this work, a molecular theory has been used to systematically study how nanoconfinement modulates the competition among these interactions and, therefore, dictates the morphology of the self-assembled layer. Two different types of confinement were considered and compared: (i) soft lateral confinement due to increasing surface coverage in a planar polyelectrolyte brush and (ii) hard vertical confinement due to the interaction of a planar polyelectrolyte brush with an opposing surface, as typically found in AFM-colloidal-tip and surface-force-apparatus experiments. It is shown that increasing the surface coverage (soft lateral confinement) or compressing the layer with an opposing wall (hard vertical confinement) have a similar qualitative effect on the morphology of the system: both types of nanoconfinement increase the stability of morphologies that extend in one or two dimensions (such as the homogeneous brush, holes and stripes) over nonextended aggregates (such as hemispherical micelles). However, vertical confinement can also lead to pillar-like structures that are not observed in the absence of the opposing wall. Interestingly, the pillar structures, which bridge the grafting and opposing surfaces, may coexist with metastable structures collapsed to the grafting surface only. This coexistence may help to understand the hysteresis commonly observed in surface-force experiments.Keywords: aggregates; molecular interactions; pH; polyelectrolyte brush; poor solvent;
Co-reporter:Rikkert J. Nap;Sung Hyun Park
Journal of Polymer Science Part B: Polymer Physics 2014 Volume 52( Issue 24) pp:1689-1699
Publication Date(Web):
DOI:10.1002/polb.23613
ABSTRACT
We have developed and applied a molecular theory that enables the investigation of the interactions between polyelectrolyte coated nanoparticles (NPs) in aqueous solutions. Potential applications of polyelectrolyte-coated NPs involve nanosensors for oil well characterization. To account for the high salinity environment encountered in an oil well or brine solution, we also considered in the theoretical description counterion condensation as well as ion–ion paring. We identified the design criteria to achieve dispersion stability of NPs coated with either polyacrylic acid (pAA), poly acrylamido-2-methylpropane sulfonate (pAMPS), or an alternating copolymer of acrylic acid and acrylamido-2-methylpropane sulfonate (pAA-a-AMPS) under brine-like conditions and quantified the effects of NP core size, molecular weight, density of the polyelectrolyte coating, and polymer chemistry. The results were summarized in stability diagrams, which predict the polymer surface coverage and molecular weight that is required for the NP solution to remain dispersed. © 2014 Wiley Periodicals, Inc. J. Polym. Sci., Part B: Polym. Phys. 2014, 52, 1689–1699
Co-reporter:Gabriel S. Longo, Monica Olvera de la Cruz, and Igal Szleifer
Langmuir 2014 Volume 30(Issue 50) pp:15335-15344
Publication Date(Web):December 1, 2014
DOI:10.1021/la5040382
We present a molecular theory to study the adsorption of different species within pH-sensitive hydrogel nanofilms. The theoretical framework allows for a molecular-level description of all the components of the system, and it explicitly accounts for the acid–base equilibrium. We concentrate on the adsorption of hexahistidine, one of the most widely used tags in bio-related systems, particularly in chromatography of proteins. The adsorption of hexahistidine within a grafted polyacid hydrogel film shows a nonmonotonic dependence on the solution pH. Depending on the salt concentration, the density of the polymer network, and the bulk concentration of peptide, substantial adsorption is predicted in the intermediate pH range where both the network and the amino acids are charged. To enhance the electrostatic attractions, the acid–base equilibrium of adsorbed hexahistidine is shifted significantly, increasing the degree of charge of the residues as compared to the bulk solution. Such a shift depends critically on the conditions of the environment at the nanoscale. At the same time, the degree of dissociation of the network becomes that of the isolated acid group in a dilute solution, which means that the network is considerably more charged than when there is no adsorbate molecules. This work provides fundamental information on the physical chemistry behind the adsorption behavior and the response of the hydrogel film. This information can be useful in designing new materials for the purification or separation/immobilization of histidine-tagged proteins.
Co-reporter:R. J. Nap and I. Szleifer
Biomaterials Science 2013 vol. 1(Issue 8) pp:814-823
Publication Date(Web):05 Apr 2013
DOI:10.1039/C3BM00181D
One of the key challenges in the development of nano carriers for drug delivery and imaging is the design of a system that selectively binds to target cells. A common strategy is to coat the delivery device with specific ligands that bind strongly to overexpressed receptors. However such devices are usually unable to discriminate between receptors found on benign and malignant cells. We demonstrate, theoretically, how one can achieve enhanced binding to target cells by using multiple physical and chemical interactions. We study the effective interactions between a polymer decorated nano micelle or nanoparticle with three types of model lipid membranes that differ in the composition of their outer leaflet. They are: (i) lipid membranes with overexpressed receptors, (ii) membranes with a given fraction of negatively charged lipids and (iii) membranes with both overexpressed receptors and negatively charged lipids. The coating contains a mixture of two short polymers, one neutral for protection and the other a polybase with a functional end-group to optimize specific binding with the overexpressed receptors and electrostatic interactions with charged lipid head-groups. The strength of the binding for the combined system is much larger than the sum of the independent electrostatic or specific interactions binding. We find a range of distances where the addition of two effective repulsive interactions become an attraction in the combined case. The changes in the strength and shape of the effective interaction are due to the coupling that exists between molecular organization, physical interactions and chemical state, e.g., protonation. The predictions provide guidelines for the design of carrier devices for targeted drug and nanoparticle delivery and give insight in the competing and highly non-additive nature of the different effective interactions in nanoscale systems in constrained environments that are ubiquitous in synthetic and biological systems.
Co-reporter:Gabriel S. Longo, Monica Olvera de la Cruz, and Igal Szleifer
ACS Nano 2013 Volume 7(Issue 3) pp:2693
Publication Date(Web):February 25, 2013
DOI:10.1021/nn400130c
Domain formation and control in pH-responsive amphiphilic polymer co-networks are studied theoretically. Two different molecular architectures of the polymer network are considered, depending on whether the pH-sensitive motif is borne by the hydrophobic or the hydrophilic monomer. When the hydrophobic polymer contains acidic groups, such chains form nanometric aggregates at acidic conditions, but they are found in a swollen state at alkaline pH. At intermediate pH, the nanoaggregation behavior of the hydrophobic polymer depends critically on the environment salt concentration. Moreover, our results indicate the presence of microphase separation into domains of swollen and aggregated hydrophobic chains. If the hydrophilic polymer is the ionizable component of the network, the nanoaggregation of hydrophobic monomers is weakly dependent on the pH and salt concentration, and except at very low volume fraction, the aggregate is the most probable conformation of the network in the entire range of pH and salt concentration studied. The two different hydrogels display quantitatively similar swelling transition and apparent pKa, but at the nanoscale, their behavior is qualitatively different. The spatial distribution of electric charge on the network as well as the local density of the different chemical species within the hydrogel can be controlled, as a function of pH and salt concentration, by the molecular architecture of the polymer network. These findings have relevance for applications in biomaterials and nanotechnology, in particular, in the design of oral delivery devices for the administration of hydrophobic drugs.Keywords: amphiphilic copolymer networks; drug delivery; hydrogels; nanoaggregation; responsive materials
Co-reporter:Mario Tagliazucchi;Martin Kröger;Orit Peleg;Yitzhak Rabin
PNAS 2013 Volume 110 (Issue 9 ) pp:3363-3368
Publication Date(Web):2013-02-26
DOI:10.1073/pnas.1212909110
The molecular structure of the yeast nuclear pore complex (NPC) and the translocation of model particles have been studied
with a molecular theory that accounts for the geometry of the pore and the sequence and anchoring position of the unfolded
domains of the nucleoporin proteins (the FG-Nups), which control selective transport through the pore. The theory explicitly
models the electrostatic, hydrophobic, steric, conformational, and acid-base properties of the FG-Nups. The electrostatic
potential within the pore, which arises from the specific charge distribution of the FG-Nups, is predicted to be negative
close to pore walls and positive along the pore axis. The positive electrostatic potential facilitates the translocation of
negatively charged particles, and the free energy barrier for translocation decreases for increasing particle hydrophobicity.
These results agree with the experimental observation that transport receptors that form complexes with hydrophilic/neutral
or positively charged proteins to transport them through the NPC are both hydrophobic and strongly negatively charged. The
molecular theory shows that the effects of electrostatic and hydrophobic interactions on the translocating potential are cooperative
and nonequivalent due to the interaction-dependent reorganization of the FG-Nups in the presence of the translocating particle.
The combination of electrostatic and hydrophobic interactions can give rise to complex translocation potentials displaying
a combination of wells and barriers, in contrast to the simple barrier potential observed for a hydrophilic/neutral translocating
particle. This work demonstrates the importance of explicitly considering the amino acid sequence and hydrophobic, electrostatic,
and steric interactions in understanding the translocation through the NPC.
Co-reporter:Mario Tagliazucchi, Yitzhak Rabin, and Igal Szleifer
ACS Nano 2013 Volume 7(Issue 10) pp:9085
Publication Date(Web):September 18, 2013
DOI:10.1021/nn403686s
This work reports a comprehensive theoretical study of the transport-rectification properties of cylindrical nanopores with neutral inner walls and chemically modified outer membrane. The chemical species on the two outer sides of the membrane have charges of opposite sign and can be either surface-confined species (i.e., surface charges) or polyelectrolyte brushes. The advantage of this design over other types of rectifying nanopores is that it requires controlling the composition of the outer walls of the pore (which are easy to access) rather than the inner walls, thus simplifying the fabrication process. Ion-current rectification in nanopores with charged outer walls is ascribed to applied-potential-induced changes in the ionic concentration within the pore. The rectification efficiency is studied as a function of pore length, radius, surface charge and bulk electrolyte concentration. An analytical model is derived for the case of surface-confined charges that predicts the current–potential curves in very good agreement with the numerical calculations. Neutral nanopores with polyelectrolyte-modified outer walls have two distinct advantages compared to surface-charged systems: (i) they exhibit higher rectification factors due to the large charge density immobilized by the polyelectrolyte brushes, and (ii) the applied potential deforms the polyelectrolyte chains toward the oppositely charged electrode. This deformation brings the polyelectrolyte brushes into the pore in the low conductivity state and expels them from the pore in the high conductivity regime. Calculations of the potentials of mean-force suggest that the applied-field-induced conformational changes can be used to control the translocation of cargoes larger than ions, such as proteins and nanoparticles.Keywords: concentration polarization; diode; electrostatics; nanochannel; nanofluidics; nonequilibrium
Co-reporter:Rikkert J. Nap, Yoonjee Park, Joyce Y. Wong, and I. Szleifer
Langmuir 2013 Volume 29(Issue 47) pp:14482-14493
Publication Date(Web):October 21, 2013
DOI:10.1021/la403143a
A molecular theoretical description is developed to describe the adsorption of nanoparticles (NPs) that are coated with polymers and functionalized with (surface) acid groups. Results are presented for the adsorption onto both negatively and positively charged surfaces as a function of pH and salt concentration, polymer coating, and NP size. An important finding is that nanoparticles that are coated with weak charge regulating acid molecules such as citric acid develop an asymmetric charge distribution close to a charged surface, due to their finite size. Depending on the sign of the surface charge of the adsorbing surface, a nanoparticle close to the surface either gains more charge or loses charge compared to its “bulk” degree of charge. This in turn influences the amount of NPs that adsorb. The effect of adsorption of negatively charged NPs onto a positively charged surface shows a nonmonotonical variation with pH. The described charging mechanism reveals that details such as size of the NP and acid distribution on the NP need to be considered to provide an accurate understanding of the adsorption process.
Co-reporter:Christopher George, Igal Szleifer, and Mark Ratner
ACS Nano 2013 Volume 7(Issue 1) pp:108
Publication Date(Web):December 2, 2012
DOI:10.1021/nn303320w
We explore the transport of electrons between electrodes that encase a two-dimensional array of metallic quantum dots linked by molecular bridges (such as α,ω alkaline dithiols). Because the molecules can move at finite temperatures, the entire transport structure comprising the quantum dots and the molecules is in dynamical motion while the charge is being transported. There are then several physical processes (physical excursions of molecules and quantum dots, electronic migration, ordinary vibrations), all of which influence electronic transport. Each can occur on a different time scale. It is therefore not appropriate to use standard approaches to this sort of electron transfer problem. Instead, we present a treatment in which three different theoretical approaches—kinetic Monte Carlo, classical molecular dynamics, and quantum transport—are all employed. In certain limits, some of the dynamical effects are unimportant. But in general, the transport seems to follow a sort of dynamic bond percolation picture, an approach originally introduced as formal models and later applied to polymer electrolytes. Different rate-determining steps occur in different limits. This approach offers a powerful scheme for dealing with multiple time scale transport problems, as will exist in many situations with several pathways through molecular arrays or even individual molecules that are dynamically disordered.Keywords: dynamic percolation; electron hopping; electron transfer; molecular conductance; quantum dots
Co-reporter:Mario Tagliazucchi and Igal Szleifer
Soft Matter 2012 vol. 8(Issue 28) pp:7292-7305
Publication Date(Web):15 May 2012
DOI:10.1039/C2SM25777G
This review discusses the properties of macromolecular layers on nano-curved surfaces, with special emphasis on the fast-growing field of polymer- and polyelectrolyte-modified nanopores and nanochannels. Organization in soft materials emerges from the competition between physical and chemical interactions acting on different length scales; the geometry of the system modulates this competition and, hence, dictates its structure and function. Theoretical studies of molecular organization in planar and curved geometries require the treatment of the coupling between interactions, chemical equilibrium and geometrical constraints. We analyse here how curvature affects the morphology of polymers on nano-curved surfaces and determines the apparent equilibrium constants of surface-confined chemical reactions, such as acid–base chemistry. We also review the practical implications of introducing soft materials into nanofluidic devices. In particular, we present and discuss recent theoretical studies and simulations of the transport of ions, solvent and large cargoes through polyelectrolyte-brush-modified pores. We believe that this basic understanding will guide experimental efforts towards the design of stimuli-responsive nanopore gates.
Co-reporter:R. J. Nap, You-Yeon Won and I. Szleifer
Soft Matter 2012 vol. 8(Issue 5) pp:1688-1700
Publication Date(Web):21 Dec 2011
DOI:10.1039/C2SM06549E
The equilibrium structures of polymers end-tethered to nanoparticles or to nanomicelles interacting with surfaces have been studied theoretically. Polymer chains chemically grafted to nanoparticles are laterally immobile. On the other hand, nanosized polymer micelles formed by polymer chains conjugated with lipids, have end-tethered chains that are laterally mobile within the self-assembled structure. Using a molecular theory, we investigated the influences of the mobile nature of the tethered chains and the nanoscale dimension of the anchoring surface on the structures and interactions of the polymers during the process of binding of the nanoparticle to a surface. We show that polymer chains with bidisperse molecular weight distributions end-tethered to a nanomicelle/nanoparticle surface segregate upon approach to a surface. The shorter chains preferentially locate in the vicinity of the surface, while the longer ones are excluded from the region between the micelle and the surface and thus become more concentrated on the opposite side of the micelle surface. The extent of this segregation is controlled by the overall surface coverage and compositions of the tethered chains, and the sizes of the short and long chains. Combining lateral mobility of the polymer tether with an end-binding capability of the chain (e.g., through a ligand–receptor interaction) can give rise to an enhancement of the interaction of the polymer nanoparticle with a surface. The results demonstrate that laterally mobility of tethered chains is an important aspect that needs to be taken into account in designing polymeric nanoparticles with enhanced surface interaction properties.
Co-reporter:Gabriel S. Longo, Monica Olvera de la Cruz and I. Szleifer
Soft Matter 2012 vol. 8(Issue 5) pp:1344-1354
Publication Date(Web):16 Dec 2011
DOI:10.1039/C1SM06708G
In this work, we extend the recently developed Molecular Theory of Weak Polyelectrolyte Gels to study hydrogel films. This approach explicitly accounts for all of the physicochemical interactions determining the thermodynamic equilibrium of these films and it incorporates molecular details of the polymer network. In particular, we applied this theoretical framework to investigate the response of a thin film of cross-linked hydrophilic polyacid chains to variations in external stimuli such as pH, salt concentration and applied electric potential. Swelling of the polyacid gel film is a continuous but sharp transition that occurs in a narrow range of bulk pH, around the pKa of the acid monomers. The width of this transition range depends on the salt concentration. The gel swells if the bulk pH is larger than the pKa of the monomers and it collapses otherwise. Swelling, however, is not due to a high degree of dissociation in the network, since the swollen gel can have very low degree of electric charge, but the result of the complex balance between the acid–base equilibrium, the gel molecular organization, and the resulting electrostatic interactions. The region close to the surface (up to a few nanometres-thick), the center of the gel, and the interface between the film and the solution have different chemical compositions, which are each different from that of the bulk solution in equilibrium with the film. In particular, the pH in all these regions can be controlled by changing the bulk pH and salt concentration. In addition, there is a gradient of pH going from the solution inside the film, whose magnitude can be tuned by varying bulk pH and salt concentration. In the region near the surface, both the pH and total charge density can be controlled by applying an electric potential. The thin film behaves as an electric insulating material. We calculated the potential of mean force for the insertion of charged nanoparticles inside the hydrogel film. Depending on the electric charge and size of the nanoparticle, there can be an attractive well or a repulsive barrier of several kBT for the nanoparticle to enter the gel from the solution. These findings are relevant in the design of a variety of functional devices using hydrogel films.
Co-reporter:Tao Wei, Marcelo A. Carignano, and Igal Szleifer
The Journal of Physical Chemistry B 2012 Volume 116(Issue 34) pp:10189-10194
Publication Date(Web):August 12, 2012
DOI:10.1021/jp304057e
In this work, we present a series of fully atomistic molecular dynamics (MD) simulations to study lysozyme’s orientation-dependent adsorption on polyethylene (PE) surface in explicit water. The simulations show that depending on the orientation of the initial approach to the surface the protein may adsorb or bounce from the surface. The protein may completely leave the surface or reorient and approach the surface resulting in adsorption. The success of the trajectory to adsorb on the surface is the result of different competing interactions, including protein–surface interactions and the hydration of the protein and the hydrophobic PE surface. The difference in the hydration of various protein sites affects the protein’s orientation-dependent behavior. Side-on orientation is most likely to result in adsorption as the protein–surface exhibits the strongest attraction. However, adsorption can also happen when lysozyme’s longest axis is tilted on the surface if the protein–surface interaction is large enough to overcome the energy barrier that results from dehydrating both the protein and the surface. Our study demonstrates the significant role of dehydration process on hydrophobic surface during protein adsorption.
Co-reporter:Jan Genzer, Shafi Arifuzzaman, Rajendra R. Bhat, Kirill Efimenko, Chun-lai Ren, and Igal Szleifer
Langmuir 2012 Volume 28(Issue 4) pp:2122-2130
Publication Date(Web):December 19, 2011
DOI:10.1021/la2038747
A combined experimental and theoretical approach establishes the long-lived nature of protein adsorption on surfaces coated with chemically grafted macromolecules. Specifically, we monitor the time dependence of adsorption of lysozyme on surfaces comprising polymer assemblies made of poly(2-hydroxyethyl methacrylate) brushes grafted onto flat silica surfaces such that they produce patterns featuring orthogonal and gradual variation of the chain length (N) and grafting density (σ). We show that in the kinetically controlled regime, the amount of adsorbed protein scales universally with the product σN, while at equilibrium the amount of adsorbed protein is governed solely by σ. Surprisingly, for moderate concentrations of protein in solution, adsorption takes more than 72 h to reach an equilibrium, or steady state. Our experimental findings are corroborated with predictions using molecular theory that provides further insight into the protein adsorption phenomenon. The theory predicts that the universal behavior observed experimentally should be applicable to polymers in poor and theta solvents and to a limited extent also to good solvent conditions. Our combined experimental and theoretical findings reveal that protein adsorption is a long-lived phenomenon, much longer than generally assumed. Our studies confirm the previously predicted important differences in behavior for the kinetic versus thermodynamic control of protein adsorption.
Co-reporter:K. H. Aaron Lau, Chunlai Ren, Sung Hyun Park, Igal Szleifer, and Phillip B. Messersmith
Langmuir 2012 Volume 28(Issue 4) pp:2288-2298
Publication Date(Web):November 22, 2011
DOI:10.1021/la203905g
Surface-grafted water-soluble polymer brushes are being intensely investigated for preventing protein adsorption to improve biomedical device function, prevent marine fouling, and enable applications in biosensing and tissue engineering. In this contribution, we present an experimental-theoretical analysis of a peptidomimetic polymer brush system with regard to the critical brush density required for preventing protein adsorption at varying chain lengths. A mussel adhesive-inspired DOPA-Lys (DOPA = 3,4-dihydroxy-phenylalanine; Lys = lysine) pentapeptide surface grafting motif enabled aqueous deposition of our peptidomimetic polypeptoid brushes over a wide range of chain densities. Critical densities of 0.88 nm–2 for a relatively short polypeptoid 10-mer to 0.42 nm–2 for a 50-mer were identified from measurements of protein adsorption. The experiments were also compared with the protein adsorption isotherms predicted by a molecular theory. Excellent agreements in terms of both the polymer brush structure and the critical chain density were obtained. Furthermore, atomic force microscopy (AFM) imaging is shown to be useful in verifying the critical brush density for preventing protein adsorption. The present coanalysis of experimental and theoretical results demonstrates the significance of characterizing the critical brush density in evaluating the performance of an antifouling polymer brush system. The high fidelity of the agreement between the experiments and molecular theory also indicate that the theoretical approach presented can aid in the practical design of antifouling polymer brush systems.
Co-reporter:Mario Tagliazucchi, Martin G. Blaber, George C. Schatz, Emily A. Weiss, and Igal Szleifer
ACS Nano 2012 Volume 6(Issue 9) pp:8397
Publication Date(Web):August 21, 2012
DOI:10.1021/nn303221y
This work presents a novel modeling approach to calculate the optical properties of gold nanoparticles coated with stimuli-responsive polymers. This approach combines, for the first time, a molecular description of the soft material with an electrodynamics calculation of the optical properties of the system. A mean-field molecular theory is first used to calculate the local density of the polymer and the position-dependent dielectric constant surrounding the nanoparticle. This information is then used to calculate the optical properties of the Au@polymer colloid by solving Maxwell’s equations for an incident electromagnetic wave. Motivated by the interest in Au@PNIPAM and Au@PVP experimental systems, the theory is applied to study the effect of polymer collapse on the position of the localized surface plasmon resonance (LSPR) of the system. The most important results of the present study are as follows: (i) the LSPR always shifts to lower energies upon polymer collapse (in agreement with experimental results); this observation implies that the red shift expected due to increasing polymer density always overcomes the blue shift expected from decreasing layer thickness; (ii) the magnitude of the LSPR shift depends nonmonotonically on surface coverage and nanoparticle radius; and (iii) the formation of aggregates on the nanoparticle surface (due to microphase segregation) decreases the magnitude of the LSPR shift. These results highlight the importance of explicitly considering the coupling between the soft material and the inorganic components in determining the optical properties of the hybrid system.Keywords: discrete dipole approximation; gold nanoparticle; localized surface plasmon resonance; LSPR sensing; Mie theory; molecular theory; stimuli-responsive polymer
Co-reporter:King Hang Aaron Lau, Chunlai Ren, Tadas S. Sileika, Sung Hyun Park, Igal Szleifer, and Phillip B. Messersmith
Langmuir 2012 Volume 28(Issue 46) pp:16099-16107
Publication Date(Web):October 26, 2012
DOI:10.1021/la302131n
Poly(N-substituted glycine) “peptoids” are a class of peptidomimetic molecules receiving significant interest as engineered biomolecules. Sarcosine (i.e., poly(N-methyl glycine)) has the simplest side chain chemical structure of this family. In this Article, we demonstrate that surface-grafted polysarcosine (PSAR) brushes exhibit excellent resistance to nonspecific protein adsorption and cell attachment. Polysarcosine was coupled to a mussel adhesive protein-inspired DOPA-Lys pentapeptide, which enabled solution grafting and control of the surface chain density of the PSAR brushes. Protein adsorption was found to decrease monotonically with increasing grafted chain densities, and protein adsorption could be completely inhibited above certain critical chain densities specific to different polysarcosine chain lengths. The dependence of protein adsorption on chain length and density was also investigated by a molecular theory. PSAR brushes at high chain length and density were shown to resist fibroblast cell attachment over a 7 week period, as well as resist the attachment of some clinically relevant bacterial strains. The excellent antifouling performance of PSAR may be related to the highly hydrophilic character of polysarcosine, which was evident from high-pressure liquid chromatography measurements of polysarcosine and water contact angle measurements of the PSAR brushes. Peptoids have been shown to resist proteolytic degradation, and polysarcosine could be produced in large quantities by N-carboxy anhydride polymerization. In summary, surface-grafted polysarcosine peptoid brushes hold great promise for antifouling applications.
Co-reporter:Mario Tagliazucchi ; Yitzhak Rabin
Journal of the American Chemical Society 2011 Volume 133(Issue 44) pp:17753-17763
Publication Date(Web):September 25, 2011
DOI:10.1021/ja2063605
Chemically modified nanopores show a strong and nontrivial coupling between ion current and the structure of the immobilized species. In this work we study theoretically the conductance and structure in polymer modified nanopores and explicitly address the problem of the coupling between ion transport and molecular organization. Our approach is based on a nonequilibrium molecular theory that couples ion conductivity with the conformational degrees of freedom of the polymer and the electrostatic and nonelectrostatic interactions among polyelectrolyte chains, ions, and solvent. We apply the theory to study a cylindrical nanopore between two reservoirs as a function of pore diameter and length, the length of the polyelectrolyte chains, their grafting density, and whether they are present or not on the outer reservoir walls. In the very low applied potential regime, where the distribution of polyelectrolyte and ions is similar to that in equilibrium, we present a simple analytical model based on the combination of the different resistances in the system that describes the conductance in excellent agreement with the calculations of the full nonequilibrium molecular theory. On the other hand, for a large applied potential bias, the theory predicts a dramatic reorganization of the polyelectrolyte chains and the ions. This reorganization results from the global optimization of the different interactions in the system under nonequilibrium conditions. For nanopores modified with long chains, this reorganization leads to two interesting physical phenomena: (i) control of polyelectrolyte morphology by the direction and magnitude of ion-fluxes and (ii) an unexpected decrease in system resistance with the applied potential bias for long chains due to the coupling between polyelectrolyte segment distribution and ion currents.
Co-reporter:Dawei Wang ; Rikkert J. Nap ; István Lagzi ; Bartlomiej Kowalczyk ; Shuangbing Han ; Bartosz A. Grzybowski
Journal of the American Chemical Society 2011 Volume 133(Issue 7) pp:2192-2197
Publication Date(Web):January 31, 2011
DOI:10.1021/ja108154a
Dissociation of ionizable ligands immobilized on nanopaticles (NPs) depends on and can be regulated by the curvature of these particles as well as the size and the concentration of counterions. The apparent acid dissociation constant (pKa) of the NP-immobilized ligands lies between that of free ligands and ligands self-assembled on a flat surface. This phenomenon is explicitly rationalized by a theoretical model that accounts fully for the molecular details (size, shape, conformation, and charge distribution) of both the NPs and the counterions.
Co-reporter:Doris Grillo, Monica Olvera de la Cruz and Igal Szleifer
Soft Matter 2011 vol. 7(Issue 10) pp:4672-4679
Publication Date(Web):14 Apr 2011
DOI:10.1039/C1SM05061C
A molecular theory is developed to assess the effect that the adsorption of a macroion onto a DPPC phospholipid bilayer has on its phase behavior. The proposed theoretical approach considers the molecular details of the phospholipid molecules, including their charge density, size and molecular conformations. It was found that the favorable electrostatic interactions between the negatively charged macroion and the zwitterionic phosphocholine head-groups lead to the stabilization of the DPPC bilayer gel phase. Consequently, the main chain transition temperature from the gel phase to the liquid-crystalline phase is raised by tens of degrees upon adsorption of the negatively charged macroion. The shift in the main transition temperature increases with the surface charge density of the adsorbed macroion. These results are in line with experimental observations and show how changes in the phospholipid bilayer environment can result in profound effects on the structural and thermodynamic behavior of the lipid films.
Co-reporter:Sung Hyun Park and Igal Szleifer
The Journal of Physical Chemistry B 2011 Volume 115(Issue 37) pp:10967-10975
Publication Date(Web):August 5, 2011
DOI:10.1021/jp2025957
All-atom molecular dynamics simulations of N-substituted glycine peptoid oligomers with methyl and methoxyethyl side chains have been carried out for chain lengths of 5, 10, 20, and 50 residues in aqueous phase at room temperature. The (ϕ, ψ) backbone dihedral angle distributions in the Ramachandran plots show that helical structures, similar to polyproline type I and type II helices, are the most favorable conformations in most peptoid oligomers studied. The left-handed helical structures are shown to be increasingly favored as the oligomer chain length grows. A significant population of cis amide bond configurations has been identified in the peptoid oligomers. By combining the analysis of ϕ and ω backbone dihedral angles, we determined the relative composition of the four major conformations favored by the backbone dihedral angles. The trans αD conformation is found to be most favored for all peptoid oligomers studies. The time correlation functions of the end-to-end distance highlight a rigid backbone structure relative to side chains for peptoid oligomers. The transition between right-handed and left-handed helical conformations is found to be very rare and between cis and trans isomerism in the amide bond completely absent in the simulation time scale. The radii of gyration for all peptoid oligomers have been found to be consistently larger in comparison to the peptide counterparts, suggesting slightly open structures for peptoids relative to peptides, whereas the fluctuations in the radius of gyration support a rigid backbone structure of peptoids.
Co-reporter:Mark J. Uline, Yitzhak Rabin, and Igal Szleifer
Langmuir 2011 Volume 27(Issue 8) pp:4679-4689
Publication Date(Web):March 22, 2011
DOI:10.1021/la104906r
Charge regulation in polyacid monolayers attached at one end to a planar surface is studied theoretically. The polyacid layers are designed to mimic single-stranded DNA monolayers. The effects of the local pH and salt concentration on the protonation states of the polyacid layer are studied using a molecular mean-field theory that includes a microscopic description of the conformations of the polyacid molecule along with electrostatic interactions, acid−base equilibrium, and excluded volume interactions. We predict that, in the case of a monovalent salt, NaCl, the amount of proton binding increases dramatically for high surface coverage of polyacid and low bulk salt concentration. When the polyelectrolyte is almost completely charge neutralized by bound protons, there is an expulsion of sodium from the layer. We show that the degree of protonation can go all the way from 0% to 100% when the bulk pH is kept fixed at 7 by changing the surface coverage of polyacid and the bulk salt concentration. The effects of increasing protonation and the expulsion of the cations from the monolayer are reduced when sodium ions are replaced by divalent magnesium ions. Our theoretical results concur with X-ray photoelectron spectroscopy studies of ssDNA monolayers on gold.
Co-reporter:Tao Wei, Marcelo A. Carignano, and Igal Szleifer
Langmuir 2011 Volume 27(Issue 19) pp:12074-12081
Publication Date(Web):August 16, 2011
DOI:10.1021/la202622s
The adsorption of lysozyme onto a polyethylene (PE) surface in an aqueous environment was investigated via molecular dynamics (MD) simulation. The adsorption can be divided into three processes: diffusion to the surface, dehydration induced by hydrophobic surface–protein interactions, and denaturation. The dehydration process is very long, around 70 ns. Structural deformations start soon after the protein reaches the surface and continue during the whole trajectory. The hydrophobic residues are slowly driven toward the surface, inducing changes in the protein’s secondary structure. The protein’s secondary structural components near the surface are more disturbed than those farther away from the surface. The lysozyme is adsorbed with its long axis parallel to the surface and displays an anisotropic mobility on the surface that is probably due to the intrinsic structure of the PE surface. Our study demonstrates the need for long-time atomistic simulation in order to gain a complete understanding of the adsorption process.
Co-reporter:Orit Peleg, Mario Tagliazucchi, Martin Kröger, Yitzhak Rabin, and Igal Szleifer
ACS Nano 2011 Volume 5(Issue 6) pp:4737
Publication Date(Web):April 27, 2011
DOI:10.1021/nn200702u
The properties of polymer layers end-grafted to the inner surface of nanopores connected to solvent reservoirs are studied theoretically as a function of solvent quality and pore geometry. Our systematic study reveals that nanoconfinement is affected by both pore radius and length and that the conformations of the polymer chains strongly depend on their grafting position along the nanopore and on the quality of the solvent. In poor solvent, polymer chains can collapse to the walls, form a compact plug in the pore, or self-assemble into domains of different shape due to microphase separation. The morphology of these domains (aggregates on pore walls or stacked micelles along the pore axis) is mainly determined by the relationship between chain length and pore radius. In other cases the number of aggregates depends on pore length. The presence of reservoirs decreases confinement at pore edges due to the changes in available volume and introduces new organization strategies not available for infinite nanochannels. In good solvent conditions, chains grafted at the pore entrances stretch out of the pore, relieving the internal osmotic pressure and increasing the entropy of the polymers. Our study also addresses the experimentally relevant case of end-grafted chains on the outer walls of the membrane surrounding the nanopore. The effect of these polymer chains on the organization within the nanopore depends on solvent quality. For good solvents the outer chains increase the confinement of the chains at the entrance of the pore; however, the effect does not result in new structures. For poor solvents the presence of the outer polymer layer may lead to changes in the morphology of the microphase-separated domains. Our results show the complex interplay between the different interactions in a confined environment and the need to develop theoretical and experimental tools for their study.Keywords: competing interactions; microphase separation; nanoconfinement; nanopore; soft matter
Co-reporter:Mario Tagliazucchi ; Omar Azzaroni
Journal of the American Chemical Society 2010 Volume 132(Issue 35) pp:12404-12411
Publication Date(Web):August 18, 2010
DOI:10.1021/ja104152g
Solid state nanochannels modified with supramolecular architectures are a new and interesting class of stimuli-responsive nanofluidic element. Their fundamental understanding requires describing the behavior of soft-materials in confined geometries and its responses to changes in solution conditions. Here, a nanochannel modified with a polyelectrolyte brush is studied with a molecular theory that incorporates the conformational behavior of the polymers, electrostatic, van der Waals, and repulsive interactions coupled with the ability of the polymer segments to regulate their charge through acid−base equilibrium. The theory predicts pH-dependent ionic conductivity in excellent agreement with experimental observations. The polymer chains undergo large conformational changes triggered by variations in the outer solution environment and the conductivity of the device is shown to be controlled by the charge state of the polymer. The degree of polymer charge is largely affected by charge regulation and nanoconfinement effects. The molecular calculations show that the apparent pKa inside the pore departs from that in solution when increasing the curvature of the nanochannel.
Co-reporter:Mark J. Uline, Shihong Meng and Igal Szleifer
Soft Matter 2010 vol. 6(Issue 21) pp:5482-5490
Publication Date(Web):16 Sep 2010
DOI:10.1039/C0SM00542H
The surface anchoring transitions of nematogens confined in thin films in the presence of surfactants at the surfaces are studied theoretically. The theoretical approach is derived from a free energy functional that includes a local Onsager type interaction as well as a microscopic description of the conformations of the surfactant molecules. The theory predicts that the effective interactions between the surfactants and the nematogens are a non-monotonic function of the surfactant area per molecule. As a result surface anchoring transitions occur from planar to homeotropic and back to planar as the surface coverage of the surfactant is increased. The location of the transitions depends on the ability of the nematogens to penetrate into the surfactant layer. The findings presented here provide a molecular picture of the role that packing plays in the interactions between flexible and rigid anisotropic molecules and how they can be used to control the phases of nematogenic thin films in devices. Furthermore, we calculate the potential of mean force on a nematogen molecule due to the presence of surfactants, confinement, and the other nematogen molecules present. The potential of mean force gives us valuable insight into the interplay of competing intermolecular forces at the surfactant-nematogen interface. The effective interactions between the surface and the nematogens can be used as one of the building blocks of field mesoscopic theories, enabling the systematic introduction of molecular information into continuous coarse-grained approaches.
Co-reporter:Sung Hyun Park, Marcelo A. Carignano, Rikkert J. Nap and Igal Szleifer
Soft Matter 2010 vol. 6(Issue 8) pp:1644-1654
Publication Date(Web):18 Feb 2010
DOI:10.1039/B923392J
We carried out molecular dynamics simulations of water droplets on self-assembled monolayers of perfluorocarbon molecules. The interactions between the water droplet and the hydrophobic fluorocarbon surface were studied by systematically changing the molecular surface coverage and the mobility of the tethered head groups of the surface chain molecules. The microscopic contact angles were determined for different fluorocarbon surface densities. The contact angle at a nanometre length scale does not show a large change with the surface density. The structure of the droplets was studied by looking at the water density profiles and water penetration near the hydrophobic surface. At surface densities near close-packed coverage of fluorocarbons, the water density shows an oscillating pattern near the boundary with a robust layered structure. As the surface density decreased and more water molecules penetrated into the fluorocarbon surface, the ordering of the water molecules at the boundary became less pronounced and the layered density structure became diffuse. The water droplet is found to induce the interfacial surface molecules to rearrange and form unique topological structures that minimize the unfavorable water–surface contacts. The local density of the fluorocarbon molecules right below the water droplet is measured to be higher than the density outside the droplet. The density difference increases as the overall surface density decreases. Two different surface morphologies emerge from the water-induced surface reorganization over the range of surface coverage explored in the study. For surface densities near close-packed monolayer coverage, the height of the fluorocarbons is maximum at the center of the droplet and minimum at the water–vapor–surface triple junction, generating a convex surface morphology under the droplet. For lower surface densities, on the other hand, the height of the fluorocarbon surface becomes maximal at and right outside the water–vapor–surface contact line and decreases quickly towards the center of the droplet, forming a concave shape of the surface. The interplay between the fluorocarbon packing and the water molecules is found to have profound consequences in many aspects of surface–water interactions, including water depletion and penetration, hydrogen bonding, and surface morphologies.
Co-reporter:Jun Soo Kim
The Journal of Physical Chemistry C 2010 Volume 114(Issue 48) pp:20864-20869
Publication Date(Web):October 12, 2010
DOI:10.1021/jp107598m
Depletion effects on the structure and interactions between polymers induced by the presence of small depleting spheres are investigated by computer simulations. As the separation between two polymers decreases, the polymers repel each other due to the loss of conformational entropy. When the polymers are immersed in a medium crowded with small depleting spheres, however, depletion attractions between the polymer segments are induced. The resulting repulsive interaction is significantly reduced when the polymer segments approach one another closer than the size of the depleting spheres. The distance-dependent potential of mean force shows a highly nonmonotonic behavior reflecting the packing of the small depleting spheres around the polymer segments. We show that the depletion potential, that is, the component of the interactions arising from the presence of the small depleting spheres, between flexible polymers is qualitatively similar to that between two large spheres. However, there are small numerical differences that arise from the connectivity of the polymer chains. We also show that Brownian dynamics simulations of a single polymer chain with depletion potential can predict polymer statistical properties in good agreement with those from molecular dynamics simulations in which depleting spheres are explicitly accounted for. Therefore, we suggest the use of the depletion potentials for computational study of crowding effects on large biopolymers such as chromatin fibers.
Co-reporter:Mario Tagliazucchi;Mónica Olvera de la Cruz
PNAS 2010 Volume 107 (Issue 12 ) pp:5300-5305
Publication Date(Web):2010-03-23
DOI:10.1073/pnas.0913340107
The competition between chemical equilibrium, for example protonation, and physical interactions determines the molecular
organization and functionality of biological and synthetic systems. Charge regulation by displacement of acid-base equilibrium
induced by changes in the local environment provides a feedback mechanism that controls the balance between electrostatic,
van der Waals, steric interactions and molecular organization. Which strategies do responsive systems follow to globally optimize
chemical equilibrium and physical interactions? We address this question by theoretically studying model layers of end-grafted
polyacids. These layers spontaneously form self-assembled aggregates, presenting domains of controlled local pH and whose
morphologies can be manipulated by the composition of the solution in contact with the film. Charge regulation stabilizes
micellar domains over a wide range of pH by reducing the local charge in the aggregate at the cost of chemical free energy
and gaining in hydrophobic interactions. This balance determines the boundaries between different aggregate morphologies.
We show that a qualitatively new form of organization arises from the coupling between physical interactions and protonation
equilibrium. This optimization strategy presents itself with polyelectrolytes coexisting in two different and well-defined
protonation states. Our results underline the need of considering the coupling between chemical equilibrium and physical interactions
due to their highly nonadditive behavior. The predictions provide guidelines for the creation of responsive polymer layers
presenting self-organized patterns with functional properties and they give insights for the understanding of competing interactions
in highly inhomogeneous and constrained environments such as those relevant in nanotechnology and those responsible for biological
cells function.
Co-reporter:Chun-lai Ren, Daniel Carvajal, Kenneth R. Shull and Igal Szleifer
Langmuir 2009 Volume 25(Issue 20) pp:12283-12292
Publication Date(Web):September 9, 2009
DOI:10.1021/la901735d
The binding of streptavidin to biotin located at the terminal ends of poly(ethylene oxide) tethered to a planar surface is studied using molecular theory. The theoretical model is applied to mimic experiments (Langmuir 2008, 24, 2472) performed using drop-shape analysis to study receptor−ligand binding at the oil/water interface. Our theoretical predictions show very good agreements with the experimental results. Furthermore, the theory enables us to study the thermodynamic and structural behavior of the PEO−biotin + streptavidin layer. The interfacial structure, shown by the volume fraction profiles of bound proteins and polymers, indicates that the proteins form a thick layer supported by stretched polymers, where the thickness of the layer is greater than the height of the protein. When the polymer spacer is composed of PEO (3000), a thick layer with multilayers of proteins is formed, supported by the stretched polymer chains. It was found that thick multilayers of proteins are formed when long spacers are present or at very high protein surface coverages on short spacers. This shows that the flexibility of the polymer spacer plays an important role in determining the structure of the bound proteins due to their ability to accommodate highly distorted conformations to optimize binding and protein interactions. Protein domains are predicted when the amount of bound proteins is small due to the existence of streptavidin−streptavidin attractive interactions. As the number of proteins is increased, the competition between attractive interactions and steric repulsions determines the stability and structure of the bound layer. The theory predicts that the competition between these two forces leads to a phase separation at higher protein concentrations. The point where this transition happens depends on both spacer length and protein surface coverage and is an important consideration for practical applications of these and other similar systems. If the goal is to maximize protein binding, it is favorable to be above the layer transition, as multiple layers can accommodate greater bound protein densities. On the other hand, if the goal is to use these bound proteins as a linker group to build more complex structures, such as when avidin or streptavidin serves as a linker between two biotinylated polymers or proteins, the optimum is to be below the layer transition such that all bound linker proteins are available for further binding.
Co-reporter:Chun-lai Ren, R. J. Nap and I. Szleifer
The Journal of Physical Chemistry B 2008 Volume 112(Issue 50) pp:16238-16248
Publication Date(Web):November 14, 2008
DOI:10.1021/jp8080904
A molecular theory to study the properties of end-tethered polymer layers, in which the polymers have the ability to form hydrogen bonds with water, is presented. The approach combines the ideas of the single-chain mean-field theory to treat tethered layers with the approach of Dormidontova ( Macromolecules, 2002, 35, 987.) to include hydrogen bonds. The generalization includes the consideration of position-dependent polymer−water and water−water hydrogen bonds. The theory is applied to model poly(ethylene oxide) (PEO), and the predictions are compared with equivalent polymer layers that do not form hydrogen bonds. It is found that increasing the temperature lowers the solubility of the PEO and results in a collapse of the layer at high enough temperatures. The properties of the layer and their temperature dependence are shown to be the result of the coupling between the conformational entropy of the chains, the ability of the polymer to form hydrogen bonds, and the intermolecular interactions. The structural and thermodynamic properties of the PEO layers, such as the lateral pressure−area isotherms and polymer chemical potentials, are studied as a function of temperature and type of tethering surface. The possibility of phase separation of the PEO layer at high enough temperature is predicted due to the reduced solubility induced by breaking of polymer−water hydrogen bonds. A discussion of the advantages and limitations of the theory, together with how to apply the approach to different hydrogen-bonding polymers, is presented.
Co-reporter:Gabriel S. Longo and David H. Thompson, I. Szleifer
Langmuir 2008 Volume 24(Issue 18) pp:10324-10333
Publication Date(Web):August 13, 2008
DOI:10.1021/la8009699
The interactions between a receptor-modified planar surface and a surface grafted with a bimodal polymer layer, where one of the polymer species is ligand functionalized, are studied using a molecular theory. The effects of changing the binding energy of the ligand−receptor pair, the polymer surface coverage, the composition, and molecular weight of both the unfunctionalized and ligand functionalized polymers on the interactions between the surfaces are investigated. Our findings show that bridging exists between the surfaces including when the molecular weight of the ligand-bearing polymer is smaller than that of the unfunctionalized polymer, even though the ligand is initially buried within the polymer layer. The distance at which the surfaces bind depends only on the molecular weight of the ligand-modified polymer, while the strength of the interaction at a given surface separation can be tuned by changing the molecular weight of the polymers, the total polymer surface coverage, and the fraction of ligated polymers. The composition of the bimodal layer alters the structure of the polymer layer, thereby influencing the strength of the steric repulsions between the surfaces. Our theoretical results show good agreement with experimental data. The present theoretical study can be used as guidelines for the design of surfaces with tailored abilities for tunning the binding strength and surface−ligand separation distances for polymer-grafted surfaces bearing specific targeting ligands.
Co-reporter:Mark J. Uline, Gabriel S. Longo, M. Schick, Igal Szleifer
Biophysical Journal (5 May 2010) Volume 98(Issue 9) pp:
Publication Date(Web):5 May 2010
DOI:10.1016/j.bpj.2010.01.036
We calculate partition coefficients of various chain anchors in liquid-ordered and liquid-disordered phases utilizing a theoretical model of a bilayer membrane containing cholesterol, dipalmitoyl phosphatidylcholine, and dioleoylphosphatidylcholine. The partition coefficients are calculated as a function of chain length, degree of saturation, and temperature. Partitioning depends on the difference between the lipid environments of the coexisting phases in which the anchors are embedded. Consequently, the partition coefficient depends on the nature of the anchor, and on the relative compositions of the coexisting phases. We find that saturated anchors prefer the denser liquid-ordered phase, and that the fraction of anchors in the liquid-ordered phase increases with increasing degree of saturation of the anchors. The partition coefficient also depends upon the location of the double bonds. Anchors with double bonds closer to the middle of the chain have a greater effect on partitioning than those near the end. Doubling the number of saturated chains increases the partitioning into the liquid-ordered phase for tails that are nearly as long or longer than those comprising the bilayer. Partitioning of such chains increases with decreasing temperature, indicating that energy considerations dominate entropic ones. In contrast, partitioning of shorter chains increases with increasing temperature, indicating that entropic considerations dominate.
Co-reporter:Rikkert J. Nap, Anže Lošdorfer Božič, Igal Szleifer, Rudolf Podgornik
Biophysical Journal (21 October 2014) Volume 107(Issue 8) pp:
Publication Date(Web):21 October 2014
DOI:10.1016/j.bpj.2014.08.032
We investigate and quantify the effects of pH and salt concentration on the charge regulation of the bacteriophage PP7 capsid. These effects are found to be extremely important and substantial, introducing qualitative changes in the charge state of the capsid such as a transition from net-positive to net-negative charge depending on the solution pH. The overall charge of the virus capsid arises as a consequence of a complicated balance with the chemical dissociation equilibrium of the amino acids and the electrostatic interaction between them, and the translational entropy of the mobile solution ions, i.e., counterion release. We show that to properly describe and predict the charging equilibrium of viral capsids in general, one needs to include molecular details as exemplified by the acid-base equilibrium of the detailed distribution of amino acids in the proteinaceous capsid shell.
Co-reporter:Gabriel S. Longo, M. Schick, I. Szleifer
Biophysical Journal (20 May 2009) Volume 96(Issue 10) pp:
Publication Date(Web):20 May 2009
DOI:10.1016/j.bpj.2009.02.043
The phase stability of a fluid lipid bilayer composed of a mixture of DC18PC, (DSPC), and a shorter DCns PC, with ns from 8 to 17, has been studied using a self-consistent field theory that explicitly includes molecular details and configurational properties of the lipid molecules. Phase separation between two liquid phases was found when there was a sufficient mismatch between the hydrophobic thicknesses of the two bilayers composed entirely of one component or the other. This occurs when ns ≤ 12 and there is a sufficient concentration of the shorter lipid. The mixture separates into a thin bilayer depleted of DSPC and a thick bilayer enriched in DSPC. Even when there is no phase separation, as in the cases when there is either insufficient concentration of a sufficiently short lipid or any concentration of a lipid with ns > 12, we observe that the effect of the shorter lipid is to increase the susceptibility of the system to fluctuations in the concentration. This is of interest, given that a common motif for the anchoring of proteins to the plasma membrane is via a myristoyl chain, that is, one with 14 carbons.
Co-reporter:Tyrone J. Yacoub, Allam S. Reddy, Igal Szleifer
Biophysical Journal (20 July 2011) Volume 101(Issue 2) pp:
Publication Date(Web):20 July 2011
DOI:10.1016/j.bpj.2011.06.015
We use molecular dynamics simulations to characterize the influence of cholesterol (Chol) on the interaction between the anticancer drug doxorubicin (DOX) and a dipalmitoyl phosphatidylcholine/Chol lipid bilayer. We calculate the potential of mean force, which gives us an estimate of the free energy barrier for DOX translocation across the membrane. We find free energy barriers of 23.1 ± 3.1 kBT, 36.8 ± 5.1 kBT, and 54.5 ± 4.7 kBT for systems composed of 0%, 15%, and 30% Chol, respectively. Our predictions agree with Arrhenius activation energies from experiments using phospholipid membranes, including 20 kBT for 0% Chol and 37.2 kBT for 20% Chol. The location of the free energy barrier for translocation across the bilayer is dependent on composition. As Chol concentration increases, this barrier changes from the release of DOX into the water to flip-flop over the membrane center. The drug greatly affects local membrane structure by attracting dipalmitoyl phosphatidylcholine headgroups, curving the membrane, and allowing water penetration. Despite its hydrophobicity, DOX facilitates water transport via its polar groups.
Co-reporter:Rikkert J. Nap, Igal Szleifer
Biophysical Journal (15 November 2008) Volume 95(Issue 10) pp:
Publication Date(Web):15 November 2008
DOI:10.1529/biophysj.108.133801
Weak polyelectrolytes tethered to cylindrical surfaces are investigated using a molecular theory. These polymers form a model system to describe the properties of aggrecan molecules, which is one of the main components of cartilage. We have studied the structural and thermodynamical properties of two interacting aggrecans with a molecular density functional theory that incorporates the acid-base equilibrium as well as the molecular properties: including conformations, size, shape, and charge distribution of all molecular species. The effect of acidity and salt concentration on the behavior is explored in detail. The repulsive interactions between two cylindrical-shaped aggrecans are strongly influenced by both the salt concentration and the pH. With increasing acidity, the polyelectrolytes of the aggrecan acquire charge and with decreasing salt concentration those charges become less screened. Consequently the interactions increase in size and range with increasing acidity and decreasing salt concentration. The size and range of the forces offers a possible explanation to the aggregation behavior of aggrecans and for their ability to resist compressive forces in cartilage. Likewise, the interdigitation of two aggrecan molecules is strongly affected by the salt concentration as well as the pH. With increasing pH, the number of charges increases, causing the repulsions between the polymers to increase, leading to a lower interdigitation of the two cylindrical polymer layers of the aggrecan molecules. The low interdigitation in charged polyelectrolytes layers provides an explanation for the good lubrication properties of polyelectrolyte layers in general and cartilage in particular.
Co-reporter:R. J. Nap and I. Szleifer
Biomaterials Science (2013-Present) 2013 - vol. 1(Issue 8) pp:NaN823-823
Publication Date(Web):2013/04/05
DOI:10.1039/C3BM00181D
One of the key challenges in the development of nano carriers for drug delivery and imaging is the design of a system that selectively binds to target cells. A common strategy is to coat the delivery device with specific ligands that bind strongly to overexpressed receptors. However such devices are usually unable to discriminate between receptors found on benign and malignant cells. We demonstrate, theoretically, how one can achieve enhanced binding to target cells by using multiple physical and chemical interactions. We study the effective interactions between a polymer decorated nano micelle or nanoparticle with three types of model lipid membranes that differ in the composition of their outer leaflet. They are: (i) lipid membranes with overexpressed receptors, (ii) membranes with a given fraction of negatively charged lipids and (iii) membranes with both overexpressed receptors and negatively charged lipids. The coating contains a mixture of two short polymers, one neutral for protection and the other a polybase with a functional end-group to optimize specific binding with the overexpressed receptors and electrostatic interactions with charged lipid head-groups. The strength of the binding for the combined system is much larger than the sum of the independent electrostatic or specific interactions binding. We find a range of distances where the addition of two effective repulsive interactions become an attraction in the combined case. The changes in the strength and shape of the effective interaction are due to the coupling that exists between molecular organization, physical interactions and chemical state, e.g., protonation. The predictions provide guidelines for the design of carrier devices for targeted drug and nanoparticle delivery and give insight in the competing and highly non-additive nature of the different effective interactions in nanoscale systems in constrained environments that are ubiquitous in synthetic and biological systems.
.jpg)







![BENZENE, [[[(3E)-4-IODO-3-METHYL-3-BUTENYL]OXY]METHYL]-](http://img.cochemist.com/ccimg/808800/808762-78-9.png)
![BENZENE, [[[(3E)-4-IODO-3-METHYL-3-BUTENYL]OXY]METHYL]-](http://img.cochemist.com/ccimg/808800/808762-78-9_b.png)




![BENZENE, [(1E)-3-METHOXY-5-METHYL-1,5-HEXADIENYL]-](http://img.cochemist.com/ccimg/538400/538367-37-2.png)
![BENZENE, [(1E)-3-METHOXY-5-METHYL-1,5-HEXADIENYL]-](http://img.cochemist.com/ccimg/538400/538367-37-2_b.png)
![Silane, [(5,5-dimethyl-1-cyclohexen-1-yl)oxy]trimethyl-](http://img.cochemist.com/ccimg/501700/501668-35-5.png)
![Silane, [(5,5-dimethyl-1-cyclohexen-1-yl)oxy]trimethyl-](http://img.cochemist.com/ccimg/501700/501668-35-5_b.png)


![Cyclohexanecarboxylicacid, 2-(1,3-dihydro-1,3-dioxo-2H-naphth[2,3-f]isoindol-2-yl)-, (1R,2R)-](http://img.cochemist.com/ccimg/446100/446044-44-6.png)
![Cyclohexanecarboxylicacid, 2-(1,3-dihydro-1,3-dioxo-2H-naphth[2,3-f]isoindol-2-yl)-, (1R,2R)-](http://img.cochemist.com/ccimg/446100/446044-44-6_b.png)
![Phosphonic acid, [(3,5-dimethoxyphenyl)methyl]-, dimethyl ester](http://img.cochemist.com/ccimg/397400/397333-50-5.png)
![Phosphonic acid, [(3,5-dimethoxyphenyl)methyl]-, dimethyl ester](http://img.cochemist.com/ccimg/397400/397333-50-5_b.png)
![Ferrocene,1-[(1R)-1-(dicyclohexylphosphino)ethyl]-2-(diphenylphosphino)-, (2S)-](http://img.cochemist.com/ccimg/360100/360048-66-4.png)
![Ferrocene,1-[(1R)-1-(dicyclohexylphosphino)ethyl]-2-(diphenylphosphino)-, (2S)-](http://img.cochemist.com/ccimg/360100/360048-66-4_b.png)












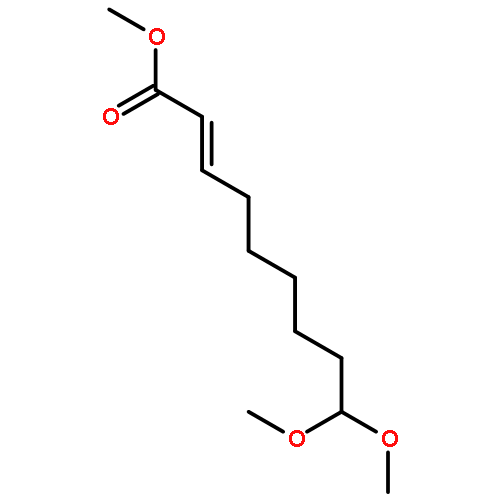

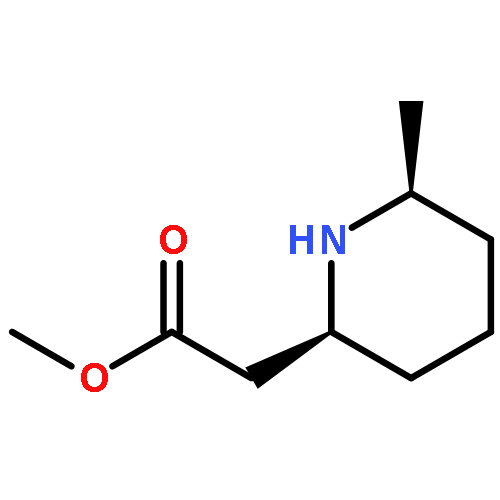






![2-Butenal, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-methyl-, (2E)-](http://img.cochemist.com/ccimg/145000/144950-82-3.png)
![2-Butenal, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-methyl-, (2E)-](http://img.cochemist.com/ccimg/145000/144950-82-3_b.png)



![Phosphonic acid, [2-(methoxymethylamino)-2-oxoethyl]-, dimethyl ester](http://img.cochemist.com/ccimg/134900/134833-70-8.png)
![Phosphonic acid, [2-(methoxymethylamino)-2-oxoethyl]-, dimethyl ester](http://img.cochemist.com/ccimg/134900/134833-70-8_b.png)



![Pentanal, 5-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/131300/131216-47-2.png)
![Pentanal, 5-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/131300/131216-47-2_b.png)








DIMETHYL-](http://img.cochemist.com/ccimg/120100/120001-35-6.png)
DIMETHYL-](http://img.cochemist.com/ccimg/120100/120001-35-6_b.png)



![Benzene, [[[2-(chloromethyl)-2-propenyl]oxy]methyl]-](http://img.cochemist.com/ccimg/114000/113964-32-2.png)
![Benzene, [[[2-(chloromethyl)-2-propenyl]oxy]methyl]-](http://img.cochemist.com/ccimg/114000/113964-32-2_b.png)









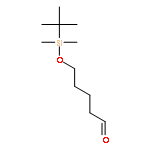
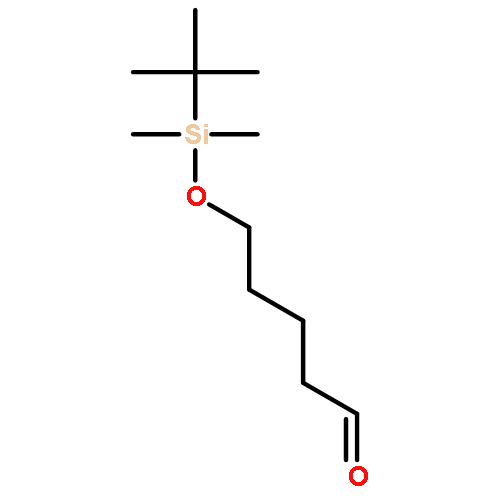
![Spiro[cyclohexane-1,4'(3'H)-[1H-7,9a]methanocyclohepta[c]pyran]-2-carboxaldehyde,hexahydro-5'-hydroxy-3,3-dimethyl-8'-methylene-1',9'-dioxo-,(1R,2R,4'aS,5'R,7'S,9'aS)-](http://img.cochemist.com/ccimg/85400/85329-59-5.png)
![Spiro[cyclohexane-1,4'(3'H)-[1H-7,9a]methanocyclohepta[c]pyran]-2-carboxaldehyde,hexahydro-5'-hydroxy-3,3-dimethyl-8'-methylene-1',9'-dioxo-,(1R,2R,4'aS,5'R,7'S,9'aS)-](http://img.cochemist.com/ccimg/85400/85329-59-5_b.png)
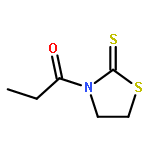

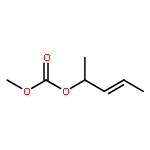
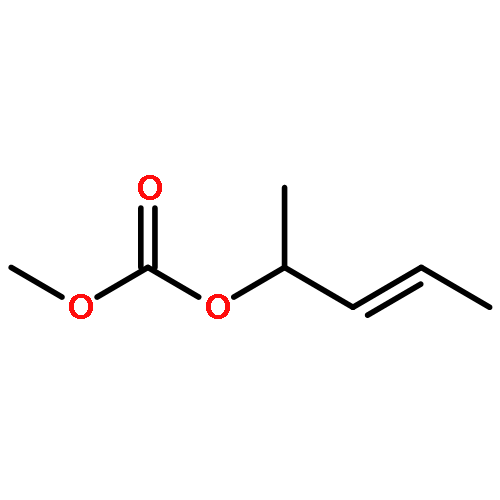

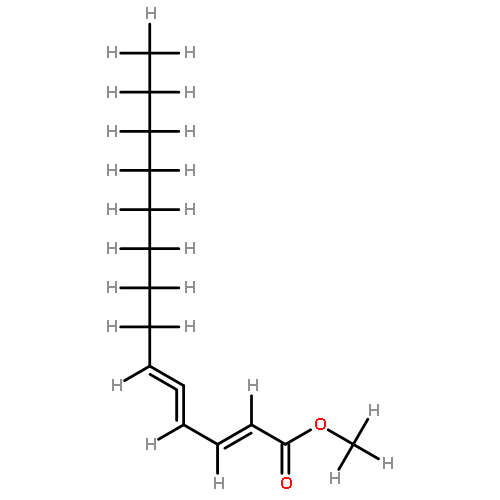
![trimethyl-[2-(trimethylsilylmethyl)prop-2-enoxy]silane](http://img.cochemist.com/ccimg/83400/83378-96-5.png)
![trimethyl-[2-(trimethylsilylmethyl)prop-2-enoxy]silane](http://img.cochemist.com/ccimg/83400/83378-96-5_b.png)
![1,7,10-Trioxadispiro[2.2.4.2]dodecane](http://img.cochemist.com/ccimg/83400/83365-44-0.png)
![1,7,10-Trioxadispiro[2.2.4.2]dodecane](http://img.cochemist.com/ccimg/83400/83365-44-0_b.png)
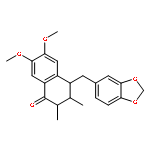

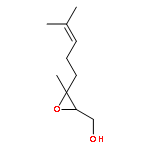
![Silane, (1,1-dimethylethyl)dimethyl[(1-methylene-2-propenyl)oxy]-](http://img.cochemist.com/ccimg/80800/80738-05-2.png)
![Silane, (1,1-dimethylethyl)dimethyl[(1-methylene-2-propenyl)oxy]-](http://img.cochemist.com/ccimg/80800/80738-05-2_b.png)




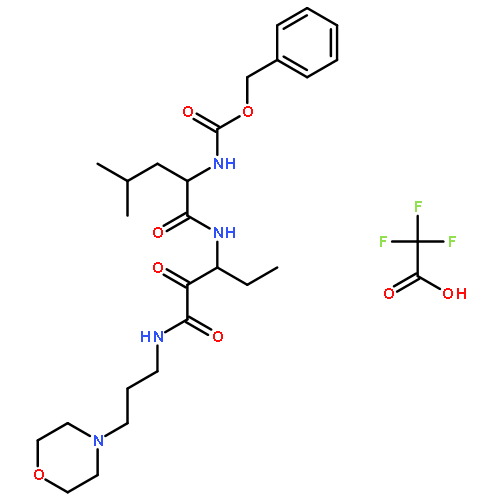






![Propanal, 2-[(tetrahydro-2H-pyran-2-yl)oxy]-, (2S)-](http://img.cochemist.com/ccimg/76500/76438-34-1.png)
![Propanal, 2-[(tetrahydro-2H-pyran-2-yl)oxy]-, (2S)-](http://img.cochemist.com/ccimg/76500/76438-34-1_b.png)




















![Silane, trimethyl[(1-methylene-4-pentenyl)oxy]-](http://img.cochemist.com/ccimg/57800/57711-32-7.png)
![Silane, trimethyl[(1-methylene-4-pentenyl)oxy]-](http://img.cochemist.com/ccimg/57800/57711-32-7_b.png)


![[1,1'-Bicyclohexyl]-2,2'-dione, (R*,S*)-](http://img.cochemist.com/ccimg/52700/52690-71-8.png)
![[1,1'-Bicyclohexyl]-2,2'-dione, (R*,S*)-](http://img.cochemist.com/ccimg/52700/52690-71-8_b.png)
![[1,1'-Bicyclopentyl]-2,2'-dione, (R*,R*)-(±)-](http://img.cochemist.com/ccimg/51900/51820-21-4.png)
![[1,1'-Bicyclopentyl]-2,2'-dione, (R*,R*)-(±)-](http://img.cochemist.com/ccimg/51900/51820-21-4_b.png)



![Ethanol, 2-[4-(1,1-dimethylethyl)cyclohexylidene]-](http://img.cochemist.com/ccimg/41500/41498-18-4.png)
![Ethanol, 2-[4-(1,1-dimethylethyl)cyclohexylidene]-](http://img.cochemist.com/ccimg/41500/41498-18-4_b.png)




![(3S)-3-[(2S,3R)-3-(acetyloxy)-1-L-valylpyrrolidin-2-yl]-3-{[N-(2-methylbutanoyl)-L-phenylalanyl]oxy}propanoic acid](http://img.cochemist.com/ccimg/37900/37878-19-6.png)
![(3S)-3-[(2S,3R)-3-(acetyloxy)-1-L-valylpyrrolidin-2-yl]-3-{[N-(2-methylbutanoyl)-L-phenylalanyl]oxy}propanoic acid](http://img.cochemist.com/ccimg/37900/37878-19-6_b.png)

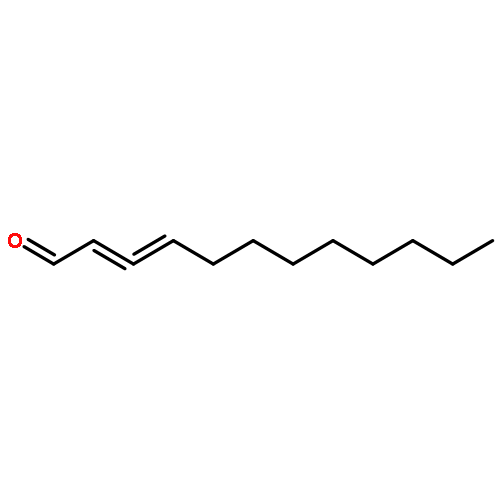











![1H-Pyrrole,2-[3-methoxy-2-(1H-pyrrol-2-ylmethylene)-2H-pyrrol-5-yl]-](http://img.cochemist.com/ccimg/24400/24311-00-0.png)
![1H-Pyrrole,2-[3-methoxy-2-(1H-pyrrol-2-ylmethylene)-2H-pyrrol-5-yl]-](http://img.cochemist.com/ccimg/24400/24311-00-0_b.png)






![Silane, [[4-(1,1-dimethylethyl)-1-cyclohexen-1-yl]oxy]trimethyl-](http://img.cochemist.com/ccimg/20000/19980-19-9.png)
![Silane, [[4-(1,1-dimethylethyl)-1-cyclohexen-1-yl]oxy]trimethyl-](http://img.cochemist.com/ccimg/20000/19980-19-9_b.png)








![[2,2'-Bi-1H-pyrrole]-5-carboxaldehyde,4-methoxy-](http://img.cochemist.com/ccimg/10500/10476-41-2.png)
![[2,2'-Bi-1H-pyrrole]-5-carboxaldehyde,4-methoxy-](http://img.cochemist.com/ccimg/10500/10476-41-2_b.png)


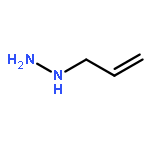
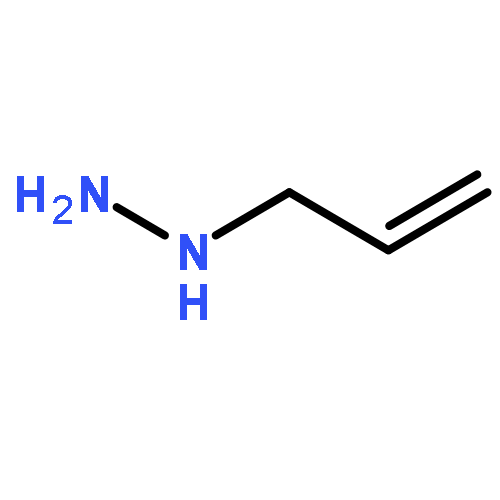
![3-methyl-4-[2-(6-methyl-piperidin-2-yl)-vinyl]-decahydro-naphtho[2,3-c]furan-1-one](http://img.cochemist.com/ccimg/6900/6879-73-8.png)
![3-methyl-4-[2-(6-methyl-piperidin-2-yl)-vinyl]-decahydro-naphtho[2,3-c]furan-1-one](http://img.cochemist.com/ccimg/6900/6879-73-8_b.png)
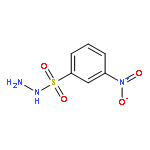
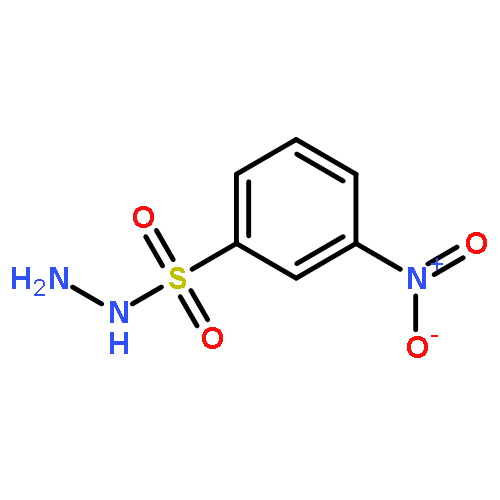

![(5S)-5r-(3,4-Dimethoxy-phenyl)-6t,7c-dimethyl-5,6,7,8-tetrahydro-naphtho[2,3-d][1,3]dioxol](http://img.cochemist.com/ccimg/4900/4892-34-6.png)
![(5S)-5r-(3,4-Dimethoxy-phenyl)-6t,7c-dimethyl-5,6,7,8-tetrahydro-naphtho[2,3-d][1,3]dioxol](http://img.cochemist.com/ccimg/4900/4892-34-6_b.png)




![Pyrrolo[1,2-a]pyrazine-1,4-dione,hexahydro-3-(phenylmethyl)-, (3S,8aS)-](http://img.cochemist.com/ccimg/3800/3705-26-8.png)
![Pyrrolo[1,2-a]pyrazine-1,4-dione,hexahydro-3-(phenylmethyl)-, (3S,8aS)-](http://img.cochemist.com/ccimg/3800/3705-26-8_b.png)





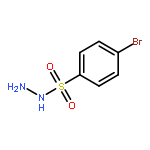
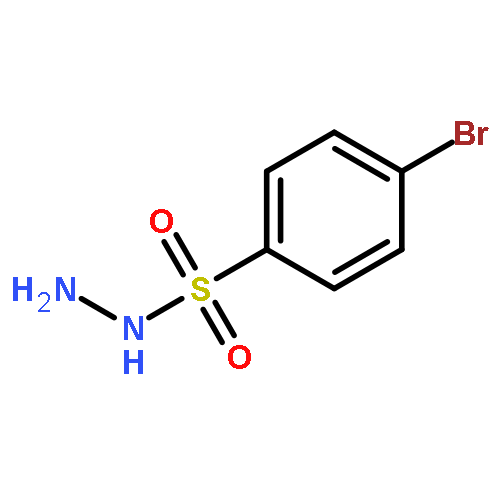


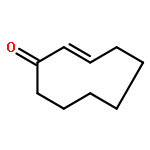
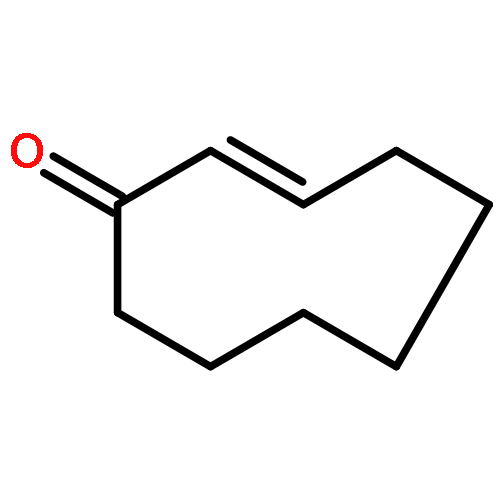













![2-Thiazolidinethione, 3-[(2R)-3,3-dimethoxy-2-methyl-1-oxopropyl]-](http://img.cochemist.com/ccimg/863400/863324-42-9.png)
![2-Thiazolidinethione, 3-[(2R)-3,3-dimethoxy-2-methyl-1-oxopropyl]-](http://img.cochemist.com/ccimg/863400/863324-42-9_b.png)















![Benzene, [(4-pentynyloxy)methyl]-](http://img.cochemist.com/ccimg/57700/57618-47-0.png)
![Benzene, [(4-pentynyloxy)methyl]-](http://img.cochemist.com/ccimg/57700/57618-47-0_b.png)
![8-BUTYL-10-[(Z)-[(5Z)-3-METHOXY-5-PYRROL-2-YLIDENEPYRROL-2-YLIDENE]METHYL]-11-AZABICYCLO[7.2.1]DODECA-1(12),9-DIENE](http://img.cochemist.com/ccimg/56300/56208-07-2.png)
![8-BUTYL-10-[(Z)-[(5Z)-3-METHOXY-5-PYRROL-2-YLIDENEPYRROL-2-YLIDENE]METHYL]-11-AZABICYCLO[7.2.1]DODECA-1(12),9-DIENE](http://img.cochemist.com/ccimg/56300/56208-07-2_b.png)
![Bicyclo[3.1.1]hept-2-ene, 6,6-dimethyl-, (1S)-](http://img.cochemist.com/ccimg/24000/23978-82-7.png)
![Bicyclo[3.1.1]hept-2-ene, 6,6-dimethyl-, (1S)-](http://img.cochemist.com/ccimg/24000/23978-82-7_b.png)
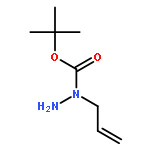
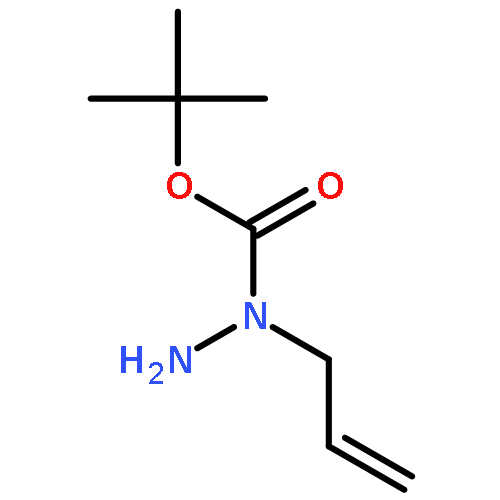
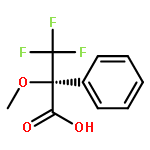
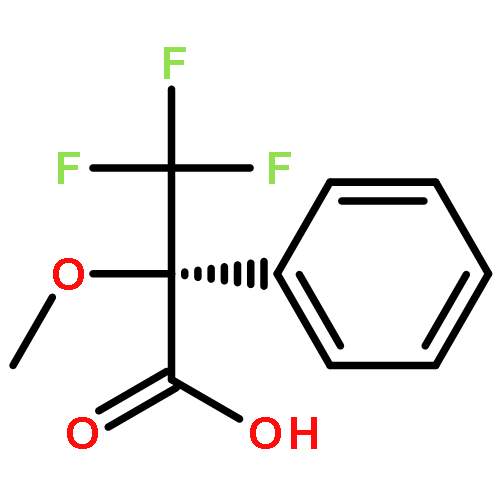


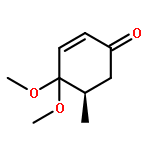
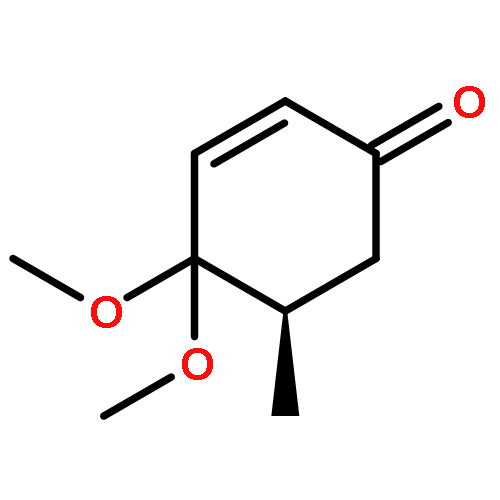



![2,2'-Bi-1H-pyrrole,4-methoxy-5-[(5-methyl-4-pentyl-2H-pyrrol-2-ylidene)methyl]-](http://img.cochemist.com/ccimg/100/82-89-3.png)
![2,2'-Bi-1H-pyrrole,4-methoxy-5-[(5-methyl-4-pentyl-2H-pyrrol-2-ylidene)methyl]-](http://img.cochemist.com/ccimg/100/82-89-3_b.png)



