Co-reporter:Matthieu Koepf;Jesse J. Bergkamp;Anne-Lucie Teillout;Manuel J. Llansola-Portoles;Gerdenis Kodis;Ana L. Moore;Thomas A. Moore
Dalton Transactions 2017 vol. 46(Issue 13) pp:4199-4208
Publication Date(Web):2017/03/27
DOI:10.1039/C6DT04647A
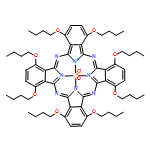
The association of different metals in stable, well-defined molecular assemblies remains a great challenge of supramolecular chemistry. In such constructs, the emergence of synergism, or cooperative effects between the different metal centers is particularly intriguing. These effects can lead to uncommon reactivity or remarkable physico-chemical properties that are not otherwise achievable. For example, the association of alkaline or alkaline-earth cations and transition metals is pivotal for the activity of several biomolecules and human-made catalysts that carry out fundamental redox transformations (water oxidation, nitrogen reduction, water–gas shift reaction, etc.). In many cases the precise nature of the interactions between the alkaline-earth cations and the redox-active transition metals remains elusive due to the difficulty of building stable molecular heterometallic assemblies that associate transition metals and alkaline or alkaline-earth cations in a controlled way. In this work we present the rational design of porphyrin-based ligands possessing a second binding site for alkaline-earth cations above the porphyrin macrocycle primary complexation site. We demonstrate that by using a combination of crown ether and carboxylic acid substituents suitably positioned on the periphery of the porphyrin, bitopic ligands can be obtained. The binding of calcium, a typical alkaline-earth cation, by the newly prepared ligands has been studied in detail and we show that a moderately large binding constant can be achieved in protic media using ligands that possess some degree of structural flexibility. The formation of Zn–Ca assemblies discussed in this work is viewed as a stepping stone towards the assembly of well defined molecular transition metal-alkaline earth bimetallic centers using a versatile organic scaffold.
Co-reporter:A. Antoniuk-Pablant, Y. Terazono, B. J. Brennan, B. D. Sherman, J. D. Megiatto, G. W. Brudvig, A. L. Moore, T. A. Moore and D. Gust
Journal of Materials Chemistry A 2016 vol. 4(Issue 8) pp:2976-2985
Publication Date(Web):16 Oct 2015
DOI:10.1039/C5TA07226C
β-Cyanoporphyrins have high positive potentials for oxidation and absorb light at longer wavelengths than most porphyrins, making them potential candidates for sensitizers in photoelectrosynthetic cells for water oxidation. In order to begin to evaluate this potential, two Zn(II) tetra-β-cyanoporphyrins have been synthesized and evaluated as sensitizers in dye sensitized solar cells using I−/I3− as the redox mediator. To prepare such specialized β-cyanoporphyrins, a new synthetic method has been developed. This approach involves reaction of Zn(CN)2 with β-brominated zinc porphyrins in the presence of tris-(dibenzylideneacetone)dipalladium. The tetra-cyanation reaction is complete under milder conditions as compared to those usually employed in previous methods and gives improved yields of up to ∼50%. The procedure allows for the cyanation of porphyrins with relatively sensitive functional groups. Examples of its application to a range of substituted tetra-arylporphyrins are reported, and the absorption and electrochemical properties of the compounds prepared are given. The results from using two of the molecules as sensitizers in dye sensitized solar cells are presented. It was found that the porphyrins produced no photocurrents in nanoparticulate TiO2-based cells, but both molecules produced photocurrents in SnO2-based cells, and are potential candidates for sensitizers in photoelectrosynthetic cells for water oxidation.
Co-reporter:Antaeres Antoniuk-Pablant, Gerdenis Kodis, Ana L. Moore, Thomas A. Moore, and Devens Gust
The Journal of Physical Chemistry B 2016 Volume 120(Issue 27) pp:6687-6697
Publication Date(Web):June 8, 2016
DOI:10.1021/acs.jpcb.6b03470
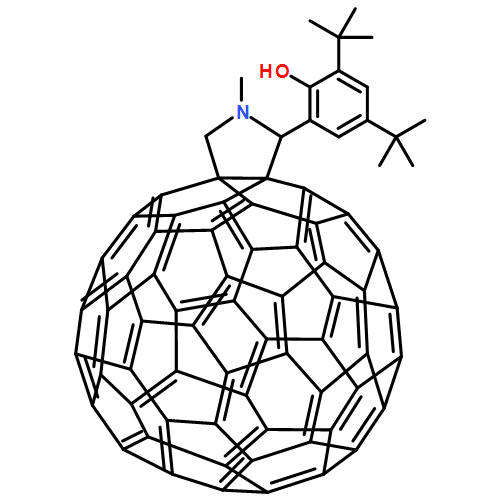
In order to investigate the possibility of a fullerene acting as an electron and/or singlet energy relay between a donor chromophore and an acceptor, a triad consisting of a fullerene (C60) covalently linked to both a porphyrin energy and electron donor (P) and a β-tetracyanoporphyrin energy and electron acceptor (CyP) was synthesized. Steady state and time-resolved spectroscopic investigations show that the porphyrin first excited singlet state donates singlet excitation and an electron to the fullerene and also donates singlet excitation to the CyP. All three processes differ in rate constant by factors of ≤1.3, and all are much faster than the decay of 1P–C60–CyP by unichromophoric processes. The fullerene excited state accepts an electron from P and donates singlet excitation energy to CyP. The P•+–C60•––CyP charge-separated state transfers an electron to CyP to produce a final P•+–C60–CyP•– state. The same state is formed from P–C60–1CyP. Overall, the final charge-separated state is formed with a quantum yield of 85% in benzonitrile, and has a lifetime of 350 ps. Rate constants for formation and quantum yields of all intermediate states were estimated from results for the triad and several model compounds. Interestingly, the intermediate P•+–C60•––CyP charge-separated state has a lifetime of 660 ps. It is longer lived than the final state in spite of stronger coupling of the radical ions. This is ascribed to the fact that recombination lies far into the inverted region of the Marcus rate constant vs thermodynamic driving force relationship.
Co-reporter:Ian Pahk, Gerdenis Kodis, Graham R. Fleming, Thomas A. Moore, Ana L. Moore, and Devens Gust
The Journal of Physical Chemistry B 2016 Volume 120(Issue 40) pp:10553-10562
Publication Date(Web):September 16, 2016
DOI:10.1021/acs.jpcb.6b07609
Nonphotochemical quenching (NPQ) is a photoprotective regulatory mechanism employed by many photosynthetic organisms to dynamically modulate energy flow within the photosynthetic apparatus in response to fluctuating light conditions. Activated by decreases in lumen pH produced during periods of high photon flux, NPQ induces rapid thermal dissipation of excess excitation energy. As a result, the rate of charge separation (CS) decreases, thereby limiting the accumulation of potentially deleterious reactive intermediates and byproducts. Herein, a molecular triad that functionally mimics the effects of NPQ associated with an artificial photosynthetic reaction center is described. Steady-state absorption and emission, time-resolved fluorescence, and transient absorption spectroscopies have been used to demonstrate a 1 order of magnitude reduction in the CS quantum yield via reversible protonation of an excited-state-quenching molecular switch moiety. As in the natural system, the populations of unquenched and quenched states and therefore the overall yields of CS were found to be dependent on acid concentration.
Co-reporter:Brian L. Watson, Benjamin D. Sherman, Ana L. Moore, Thomas A. Moore and Devens Gust
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 24) pp:15788-15796
Publication Date(Web):18 May 2015
DOI:10.1039/C5CP00860C
A new sensitizer motif for dye sensitized solar cells (DSSC) has been developed. A heteroaromatic moiety containing a pyrazine ring links two porphyrin chromophores to the metal oxide surface via two carboxylic acid attachment groups. A test DSSC sensitized with the new molecule was 3.5 times more efficient than a similar cell sensitized by a single porphyrin model compound. The open circuit photovoltage was increased by a modest factor of 1.3, but the photocurrent increased by a factor of 2.7. Most of the increase is attributed to a reduced rate of charge recombination of the charge separated state formed by photoinduced electron transfer from the excited sensitizer to the TiO2, although some of the difference is due to increased light absorption resulting from more dye on the photoanode. Increased light absorption due to the pyrazine-containing group may also play a role. The design illustrated here could also be used to link complementary sensitizers or antenna moieties in order to increase spectral coverage.
Co-reporter:Yuichi Terazono; Gerdenis Kodis; Mirianas Chachisvilis; Brian R. Cherry; Maxime Fournier; Ana Moore; Thomas A. Moore
Journal of the American Chemical Society 2014 Volume 137(Issue 1) pp:245-258
Publication Date(Web):December 16, 2014
DOI:10.1021/ja510267c
A recently reported synthetic method has been employed to prepare several arrays of free base and zinc porphyrins. In the arrays, the porphyrins are arranged around a central benzene ring. The lack of aryl rings in the linkages to the central benzene ring, coupled with the presence of only one meso-aryl substituent on each porphyrin, allows strong electronic interactions between the porphyrin macrocycles. In arrays containing two or six porphyrins, a variety of evidence indicates that the porphyrins exist as twist-stacked dimers reminiscent of the special pairs of bacteriochlorophylls found in some photosynthetic bacteria. These dimers feature van der Waals contact between the macrocycles, and demonstrate excitonic splitting due to π–π interactions. The excitonic effects split and blue-shift the Soret absorptions, and slightly broaden the Q-band absorptions and shift them to longer wavelengths. The interactions also lower the first oxidation potentials by ca. 100 mV, and the arrays show evidence for delocalization of the radical cation over both porphyrins in the dimer. The arrays demonstrate singlet–singlet energy transfer among the chromophores. Arrays of this type will be good models for some aspects of the interactions of photosynthetic pigments, including those of reaction center special pairs and possibly quantum coherence effects. They can also be useful in artificial photosynthetic constructs.
Co-reporter:Graeme Copley ; Jason G. Gillmore ; Jeffrey Crisman ; Gerdenis Kodis ; Christopher L. Gray ; Brian R. Cherry ; Benjamin D. Sherman ; Paul A. Liddell ; Michelle M. Paquette ; Laimonas Kelbauskas ; Natia L. Frank ; Ana L. Moore ; Thomas A. Moore
Journal of the American Chemical Society 2014 Volume 136(Issue 34) pp:11994-12003
Publication Date(Web):July 29, 2014
DOI:10.1021/ja504879p
Two molecules in which the intensity of shorter-wavelength fluorescence from a strong fluorophore is modulated by longer-wavelength irradiation of an attached merocyanine–spirooxazine reverse photochromic moiety have been synthesized and studied. This unusual fluorescence behavior is the result of quenching of fluorophore fluorescence by the thermally stable, open, zwitterionic form of the spirooxazine, whereas the photogenerated closed, spirocyclic form has no effect on the fluorophore excited state. The population ratio of the closed and open forms of the spirooxazine is controlled by the intensity of the longer-wavelength modulated light. Both square wave and sine wave modulation were investigated. Because the merocyanine–spirooxazine is an unusual reverse photochrome with a thermally stable long-wavelength absorbing form and a short-wavelength absorbing photogenerated isomer with a very short lifetime, this phenomenon does not require irradiation of the molecules with potentially damaging ultraviolet light, and rapid modulation of fluorescence is possible. Molecules demonstrating these properties may be useful in fluorescent probes, as their use can discriminate between probe fluorescence and various types of adventitious “autofluorescence” from other molecules in the system being studied.
Co-reporter:Robert A. Schmitz, Paul A. Liddell, Gerdenis Kodis, Michael J. Kenney, Bradley J. Brennan, Nolan V. Oster, Thomas A. Moore, Ana L. Moore and Devens Gust
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 33) pp:17569-17579
Publication Date(Web):04 Jul 2014
DOI:10.1039/C4CP02105C
A semiconducting porphyrin polymer that is solution processable and soluble in organic solvents has been synthesized, and its spectroscopic and electrochemical properties have been investigated. The polymer consists of diarylporphyrin units that are linked at meso-positions by aminophenyl groups, thus making the porphyrin rings an integral part of the polymer backbone. Hexyl chains on two of the aryl groups impart solubility. The porphyrin units interact only weakly in the ground electronic state. Excitation produces a local excited state that rapidly evolves into a state with charge-transfer character (CT) involving the amino nitrogen and the porphyrin macrocycle. Singlet excitation energy is transferred between porphyrin units in the chain with a time constant of ca. 210 ps. The final CT state has a lifetime of several nanoseconds, and the first oxidation of the polymer occurs at ca. 0.58 V vs. SCE. These properties make the polymer a suitable potential excited state electron donor to a variety of fullerenes or other acceptor species, suggesting that the polymer may find use in organic photovoltaics, sensors, and similar applications.
Co-reporter:Bradley J. Brennan, Manuel J. Llansola Portolés, Paul A. Liddell, Thomas A. Moore, Ana L. Moore and Devens Gust
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 39) pp:16605-16614
Publication Date(Web):25 Jul 2013
DOI:10.1039/C3CP52156G
A tetra-arylporphyrin dye was functionalized with three different anchoring groups used to attach molecules to metal oxide surfaces. The physical, photophysical and electrochemical properties of the derivatized porphyrins were studied, and the dyes were then linked to mesoporous TiO2. The anchoring groups were β-vinyl groups bearing either a carboxylate, a phosphonate or a siloxy moiety. The siloxy linkages were made by treatment of the metal oxide with a silatrane derivative of the porphyrin. The surface binding and lability of the anchored molecules were studied, and dye performance was compared in a dye-sensitized solar cell (DSSC). Transient absorption spectroscopy was used to study charge recombination processes. At comparable surface concentration, the porphyrin showed comparable performance in the DSSC, regardless of the linker. However, the total surface coverage achievable with the carboxylate was about twice that obtainable with the other two linkers, and this led to higher current densities for the carboxylate DSSC. On the other hand, the carboxylate-linked dyes were readily leached from the metal oxide surface under alkaline conditions. The phosphonates were considerably less labile, and the siloxy-linked porphyrins were most resistant to leaching from the surface. The use of silatrane proved to be a practical and convenient way to introduce the siloxy linkages, which can confer greatly increased stability on dye-sensitized electrodes with photoelectrochemical performance comparable to that of the other linkers.
Co-reporter:Bradley J. Brennan, Paul A. Liddell, Thomas A. Moore, Ana L. Moore, and Devens Gust
The Journal of Physical Chemistry B 2013 Volume 117(Issue 1) pp:426-432
Publication Date(Web):December 10, 2012
DOI:10.1021/jp3099945
Charge transport within films of several new types of electropolymerized porphyrin and porphyrin-fullerene dyad polymers was studied in order to obtain information on the suitability of these organic semiconductors for applications in solar energy conversion, sensor devices, etc. The films, prepared by electropolymerization on a conductive substrate, were immersed in acetonitrile and studied using chronocoulometric and cyclic voltammetric electrochemical methods. The charge diffusion coefficients were found to be dependent upon the electrolytic medium. Electrolyte anion size plays a significant role in determining the rate of migration of charge through the polymers, demonstrating that migration of positive charge is accompanied by migration of negative counterions. Bulkier anions markedly decrease the charge diffusion coefficient. This strong dependence suggests that anion mobility is the rate-limiting process for diffusional charge transport within the porphyrin polymer films and that the largest rates obtained are lower limits to the intrinsic cation mobility. With electrolytes containing the relatively small perchlorate anion, charge diffusion coefficients of the porphyrin polymers were similar to those reported for polyaniline under acidic conditions. The charge diffusion coefficient for a zinc porphyrin polymer was found to decrease 2 orders of magnitude in the presence of pyridine, suggesting that metal-containing porphyrins polymer films may have sensor applications. Cation (hole) mobilities previously reported in the literature for porphyrin-containing polymers with chemical structures quite different from those investigated here were much smaller than those found for the polymers in this study, but further investigation suggests that the differences are due to choice of electrode size and material.
Co-reporter:Vikas Garg, Gerdenis Kodis, Paul A. Liddell, Yuichi Terazono, Thomas A. Moore, Ana L. Moore, and Devens Gust
The Journal of Physical Chemistry B 2013 Volume 117(Issue 38) pp:11299-11308
Publication Date(Web):March 27, 2013
DOI:10.1021/jp402265e
In photosynthesis, sunlight is absorbed mainly by antenna chromophores that transfer singlet excitation energy to reaction centers for conversion to useful electrochemical energy. Antennas may likewise be useful in artificial photosynthetic systems that use sunlight to make fuels or electricity. Here, we report the synthesis and spectroscopic properties of a molecular hexad comprising two porphyrin moieties and four coumarin antenna chromophores, all organized by a central hexaphenylbenzene core. Light absorbed by any of the coumarins is transferred to a porphyrin on the 1–10 ps time scale, depending on the site of initial excitation. The quantum yield of singlet energy transfer is 1.0. The energy transfer rate constants are consistent with transfer by the Förster dipole–dipole mechanism. A pyridyl-bearing fullerene moiety self-assembles to the form of the hexad containing zinc porphyrins to yield an antenna–reaction center complex. In the resulting heptad, energy transfer to the porphyrins is followed by photoinduced electron transfer to the fullerene with a time constant of 3 ps. The resulting P•+–C60•– charge-separated state is formed with an overall quantum yield of 1.0 and decays with a time constant of 230 ps in 1,2-difluorobenzene as the solvent.
Co-reporter:Julien Frey, Gerdenis Kodis, Stephen D. Straight, Thomas A. Moore, Ana L. Moore, and Devens Gust
The Journal of Physical Chemistry A 2013 Volume 117(Issue 3) pp:607-615
Publication Date(Web):December 21, 2012
DOI:10.1021/jp3106887
Photochromes may be reversibly photoisomerized between two metastable states and their properties can influence, and be influenced by, other chromophores in the same molecule through energy or electron transfer. In the photochemically active molecular tetrad described here, a porphyrin has been covalently linked to a fullerene electron acceptor, a quinoline-derived dihydroindolizine photochrome, and a dithienylethene photochrome. The porphyrin first excited singlet state undergoes photoinduced electron transfer to the fullerene to generate a charge-separated state. The quantum yield of charge separation is modulated by the two photochromes: one isomer of each quenches the porphyrin excited state, reducing the quantum yield of electron transfer to near zero. Interestingly, when the molecule is illuminated with white light, the quantum yield decreases as the white light intensity is increased, generating an out-of-phase response of the quantum yield to white light. However, when the same experiment is performed in the presence of additional, steady-state UV illumination, a phase inversion occurs. The quantum yield of electron transfer now increases with increasing white light intensity. Such effects illustrate emergent complexity in a relatively simple system and could find applications in molecular logic, photochemical labeling and drug delivery, and photoprotection for artificial photosynthetic molecules. The photochemistry leading to this behavior is discussed.
Co-reporter:Devens Gust, Joakim Andréasson, Uwe Pischel, Thomas A. Moore and Ana L. Moore
Chemical Communications 2012 vol. 48(Issue 14) pp:1947-1957
Publication Date(Web):05 Dec 2011
DOI:10.1039/C1CC15329C
Photochromes are chromophores that are reversibly isomerized between two metastable forms using light, or light and heat. When photochromes are covalently linked to other chromophores, they can act as molecular photonic analogues of electronic transistors. As bistable switches, they can be incorporated into the design of molecules capable of binary arithmetic and both combinatorial and sequential digital logic operations. Small ensembles of such molecules can perform analogue signal modulation similar to that carried out by transistor amplifiers. Examples of molecules that perform multiple logic functions, act as control elements for fluorescent reporters, and mimic natural photoregulatory functions are presented.
Co-reporter:Yuichi Terazono, Emily J. North, Ana L. Moore, Thomas A. Moore, and Devens Gust
Organic Letters 2012 Volume 14(Issue 7) pp:1776-1779
Publication Date(Web):March 15, 2012
DOI:10.1021/ol300267j
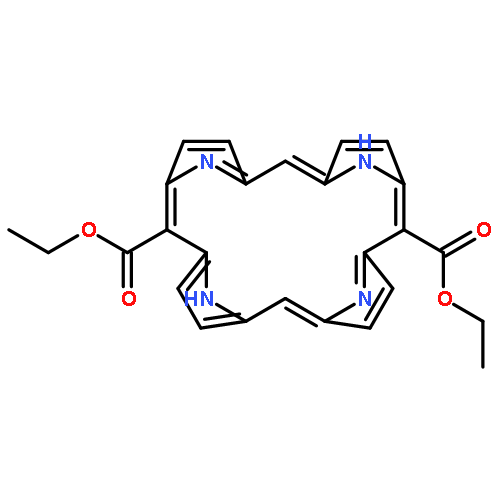
A variant of the MacDonald approach was devised for the synthesis of porphyrins. A new base-catalyzed one-step synthesis of 1,9-dipyrromethane–dicarbinols was achieved by Friedel–Crafts alkylation of dipyrromethanes using commercially available ethyl glyoxylate solution in toluene. This method avoids the use of acid chlorides, Grignard reagents, borohydride reductions, and acidic conditions. The [2 + 2] condensation of dipyrromethanedicarbinols and dipyrromethanes yielded 5,15-di(ethoxycarbonyl)porphyrins.
Co-reporter:Gary F. Moore, Jackson D. Megiatto, Michael Hambourger, Miguel Gervaldo, Gerdenis Kodis, Thomas A. Moore, Devens Gust and Ana L. Moore
Photochemical & Photobiological Sciences 2012 vol. 11(Issue 6) pp:1018-1025
Publication Date(Web):23 Feb 2012
DOI:10.1039/C2PP05351A
We report the photophysical and electrochemical properties of phenol–pyrrolidino[60]fullerenes 1 and 2, in which the phenol hydroxyl group is ortho and para to the pyrrolidino group, respectively, as well as those of a phenyl–pyrrolidino[60]fullerene model compound, 3. For the ortho analog 1, the presence of an intramolecular hydrogen bond is supported by 1H NMR and FTIR characterization. The redox potential of the phenoxyl radical–phenol couple in this architecture is 240 mV lower than that observed in the associated para compound 2. Further, the C60 excited-state lifetime of the hydrogen-bonded compound 1 in benzonitrile is 260 ps, while the corresponding lifetime for 2 is identical to that of the model compound 3 at 1.34 ns. Addition of excess organic acid to a benzonitrile solution of 1 gives rise to a new species, 4, with an excited-state lifetime of 1.40 ns. In nonpolar aprotic solvents such as toluene, all three compounds have a C60 excited-state lifetime of ∼1 ns. These results suggest that the presence of an intramolecular H-bond in 1 poises the potential of phenoxyl radical–phenol redox couple at a value that it is thermodynamically capable of reducing the photoexcited fullerene. This is not the case for the para analog 2 nor is it the case for the protonated species 4. This work illustrates that in addition to being used as light activated electron acceptors, pyrrolidino fullerenes are also capable of acting as built-in proton-accepting units that influence the potential of an attached donor when organized in an appropriate molecular design.
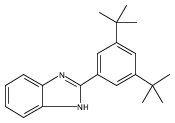
Co-reporter:Antaeres Antoniuk-Pablant;Jackson D. Megiatto, Jr.;Benjamin D. Sherman;Miguel Gervaldo;Gerdenis Kodis;Ana L. Moore;Thomas A. Moore
PNAS 2012 Volume 109 (Issue 39 ) pp:
Publication Date(Web):2012-09-25
DOI:10.1073/pnas.1118348109
In the photosynthetic photosystem II, electrons are transferred from the manganese-containing oxygen evolving complex (OEC)
to the oxidized primary electron-donor chlorophyll P680•+ by a proton-coupled electron transfer process involving a tyrosine-histidine pair. Proton transfer from the tyrosine phenolic
group to a histidine nitrogen positions the redox potential of the tyrosine between those of P680•+ and the OEC. We report the synthesis and time-resolved spectroscopic study of a molecular triad that models this electron
transfer. The triad consists of a high-potential porphyrin bearing two pentafluorophenyl groups (PF10), a tetracyanoporphyrin electron acceptor (TCNP), and a benzimidazole-phenol secondary electron-donor (Bi-PhOH). Excitation
of PF10 in benzonitrile is followed by singlet energy transfer to TCNP (τ = 41 ps), whose excited state decays by photoinduced electron transfer (τ = 830 ps) to yield . A second electron transfer reaction follows (τ < 12 ps), giving a final state postulated as BiH+-PhO•-PF10-TCNP•-, in which the phenolic proton now resides on benzimidazole. This final state decays with a time constant of 3.8 μs. The triad
thus functionally mimics the electron transfers involving the tyrosine-histidine pair in PSII. The final charge-separated
state is thermodynamically capable of water oxidation, and its long lifetime suggests the possibility of coupling systems
such as this system to water oxidation catalysts for use in artificial photosynthetic fuel production.
Co-reporter:Vikas Garg ; Gerdenis Kodis ; Mirianas Chachisvilis ; Michael Hambourger ; Ana L. Moore ; Thomas A. Moore
Journal of the American Chemical Society 2011 Volume 133(Issue 9) pp:2944-2954
Publication Date(Web):February 14, 2011
DOI:10.1021/ja1083078
Photosynthetic reaction centers convert excitation energy from absorbed sunlight into chemical potential energy in the form of a charge-separated state. The rates of the electron transfer reactions necessary to achieve long-lived, high-energy charge-separated states with high quantum yields are determined in part by precise control of the electronic coupling among the chromophores, donors, and acceptors and of the reaction energetics. Successful artificial photosynthetic reaction centers for solar energy conversion have similar requirements. Control of electronic coupling in particular necessitates chemical linkages between active component moieties that both mediate coupling and restrict conformational mobility so that only spatial arrangements that promote favorable coupling are populated. Toward this end, we report the synthesis, structure, and photochemical properties of an artificial reaction center containing two porphyrin electron donor moieties and a fullerene electron acceptor in a macrocyclic arrangement involving a ring of 42 atoms. The two porphyrins are closely spaced, in an arrangement reminiscent of that of the special pair in bacterial reaction centers. The molecule is produced by an unusual cyclization reaction that yields mainly a product with C2 symmetry and trans-2 disubstitution at the fullerene. The macrocycle maintains a rigid, highly constrained structure that was determined by UV−vis spectroscopy, NMR, mass spectrometry, and molecular modeling at the semiempirical PM6 and DFT (B3LYP/6-31G**) levels. Transient absorption results for the macrocycle in 2-methyltetrahydrofuran reveal photoinduced electron transfer from the porphyrin first excited singlet state to the fullerene to form a P•+−C60•−−P charge separated state with a time constant of 1.1 ps. Photoinduced electron transfer to the fullerene excited singlet state to form the same charge-separated state has a time constant of 15 ps. The charge-separated state is formed with a quantum yield of essentially unity and has a lifetime of 2.7 ns. The ultrafast charge separation coupled with charge recombination that is over 2000 times slower is consistent with a very rigid molecular structure having a small reorganization energy for electron transfer, relative to related porphyrin−fullerene molecules.
Co-reporter:Yuichi Terazono ; Gerdenis Kodis ; Kul Bhushan ; Julia Zaks ; Christopher Madden ; Ana L. Moore ; Thomas A. Moore ; Graham R. Fleming
Journal of the American Chemical Society 2011 Volume 133(Issue 9) pp:2916-2922
Publication Date(Web):February 11, 2011
DOI:10.1021/ja107753f
One mechanism used by plants to protect against damage from excess sunlight is called nonphotochemical quenching (NPQ). Triggered by low pH in the thylakoid lumen, NPQ leads to conversion of excess excitation energy in the antenna system to heat before it can initiate production of harmful chemical species by photosynthetic reaction centers. Here we report a synthetic hexad molecule that functionally mimics the role of the antenna in NPQ. When the hexad is dissolved in an organic solvent, five zinc porphyrin antenna moieties absorb light, exchange excitation energy, and ultimately decay by normal photophysical processes. Their excited-state lifetimes are long enough to permit harvesting of the excitation energy for photoinduced charge separation or other work. However, when acid is added, a pH-sensitive dye moiety is converted to a form that rapidly quenches the first excited singlet states of all five porphyrins, converting the excitation energy to heat and rendering the porphyrins kinetically incompetent to readily perform useful photochemistry.
Co-reporter:Tse-Luen Wee ; Benjamin D. Sherman ; Devens Gust ; Ana L. Moore ; Thomas A. Moore ; Yun Liu ;Juan C. Scaiano
Journal of the American Chemical Society 2011 Volume 133(Issue 42) pp:16742-16745
Publication Date(Web):September 26, 2011
DOI:10.1021/ja206280g
New cobalt-based nanocomposites have been prepared by photoreduction of Co2+ salts to generate cobalt nanoparticles deposited on carbon-based materials such as nanocyrstalline diamond and carbon felt. Spontaneous air oxidation converts the metal to Co2O3 which has been tested as a water oxidation catalyst. This work demonstrates that the cobalt oxide nanostructures can be deposited on various carbon surfaces and can catalyze the four-electron oxidation of water to oxygen under anodic bias.
Co-reporter:Joakim Andréasson, Uwe Pischel, Stephen D. Straight, Thomas A. Moore, Ana L. Moore, and Devens Gust
Journal of the American Chemical Society 2011 Volume 133(Issue 30) pp:11641-11648
Publication Date(Web):May 12, 2011
DOI:10.1021/ja203456h
Photochromes are photoswitchable, bistable chromophores which, like transistors, can implement binary logic operations. When several photochromes are combined in one molecule, interactions between them such as energy and electron transfer allow design of simple Boolean logic gates and more complex logic devices with all-photonic inputs and outputs. Selective isomerization of individual photochromes can be achieved using light of different wavelengths, and logic outputs can employ absorption and emission properties at different wavelengths, thus allowing a single molecular species to perform several different functions, even simultaneously. Here, we report a molecule consisting of three linked photochromes that can be configured as AND, XOR, INH, half-adder, half-subtractor, multiplexer, demultiplexer, encoder, decoder, keypad lock, and logically reversible transfer gate logic devices, all with a common initial state. The system demonstrates the advantages of light-responsive molecules as multifunctional, reconfigurable nanoscale logic devices that represent an approach to true molecular information processing units.
Co-reporter:Bradley J. Brennan, Michael J. Kenney, Paul A. Liddell, Brian R. Cherry, Jian Li, Ana L. Moore, Thomas A. Moore and Devens Gust
Chemical Communications 2011 vol. 47(Issue 36) pp:10034-10036
Publication Date(Web):09 Aug 2011
DOI:10.1039/C1CC13596A
A method for radical coupling of porphyrins using copper(II) salts as one-electron oxidants was developed. A Zn(II)-porphyrin bearing an aminophenyl group yielded porphyrin oligomers, and two tri-arylporphyrins were oxidized to form doubly and triply linked dimers. Bromination of doubly linked dimers gave macrocycles with twisted skeletons.
Co-reporter:Amy E. Keirstead ; James W. Bridgewater ; Yuichi Terazono ; Gerdenis Kodis ; Stephen Straight ; Paul A. Liddell ; Ana L. Moore ; Thomas A. Moore
Journal of the American Chemical Society 2010 Volume 132(Issue 18) pp:6588-6595
Publication Date(Web):April 21, 2010
DOI:10.1021/ja1019595
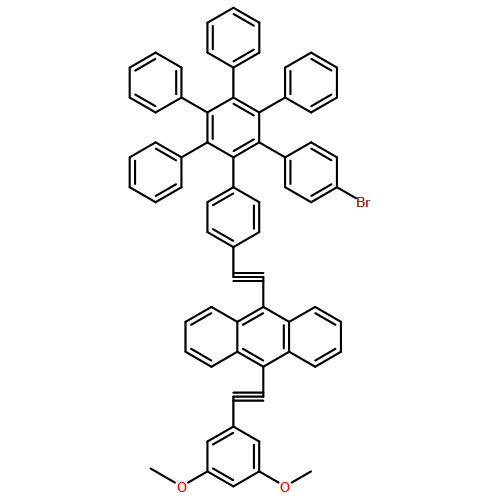
A molecular “hexad” in which five bis(phenylethynyl)anthracene (BPEA) fluorophores and a dithienylethene photochrome are organized by a central hexaphenylbenzene unit has been prepared. Singlet−singlet energy transfer among the BPEA units occurs on the 0.4 and 60 ps time scales, and when the dithienylethene is in the open form, the BPEA units fluoresce in the 515 nm region with a quantum yield near unity. When the dithienylethene is photoisomerized by UV light to the closed form, which absorbs in the 500−700 nm region, the closed isomer strongly quenches all of the excited singlet states of BPEA via energy transfer, causing the fluorescence quantum yield to drop to near zero. This photochemical behavior permits the hexad to function in a manner analogous to a triode vacuum tube or transistor. When a solution of the hexad is irradiated with steady-state light at 350 nm and with red light (>610 nm) of modulated intensity, the BPEA fluorescence excited by the 350 nm light is modulated accordingly. The fluorescence corresponds to the output of a triode tube or transistor and the modulated red light to the grid signal of the tube or gate voltage of the transistor. Frequency modulation, amplitude modulation, and phase modulation are all observed. The unusual ability to modulate intense, shorter-wavelength fluorescence with longer-wavelength light could be useful for the detection of fluorescence from probe molecules without interference from other emitters in biomolecular or nanotechnological applications.
Co-reporter:Miguel Gervaldo, Paul A. Liddell, Gerdenis Kodis, Bradley J. Brennan, Christopher R. Johnson, James W. Bridgewater, Ana L. Moore, Thomas A. Moore and Devens Gust
Photochemical & Photobiological Sciences 2010 vol. 9(Issue 7) pp:890-900
Publication Date(Web):01 Apr 2010
DOI:10.1039/C0PP00013B
A hole- and electron-conducting polymer has been prepared by electropolymerization of a porphyrin–fullerene monomer. The porphyrin units are linked by aminophenyl groups to form a linear chain in which the porphyrin is an integral part of the polymer backbone. The absorption spectrum of a film formed on indium-tin-oxide-coated glass resembles that of a model porphyrin–fullerene dyad, but with significant peak broadening. The film demonstrates a first oxidation potential of 0.75 V vs. SCE, corresponding to oxidation of the porphyrin polymer, and a first reduction potential of −0.63 V vs. SCE, corresponding to fullerene reduction. Time-resolved fluorescence studies show that the porphyrin first excited singlet state is strongly quenched by photoinduced electron transfer to fullerene. Transient absorption investigations reveal that excitation generates mobile charge carriers that recombine by both geminate and nongeminate pathways over a large range of time scales. Similar studies on a related polymer that lacks the fullerene component show complex, laser-intensity-dependent photoinduced electron transfer behavior. The properties of the porphyrin–fullerene electropolymer suggest that it may be useful in organic photovoltaic applications, wherein light absorption leads to charge separation within picoseconds in a “molecular heterojunction” with no requirement for exciton migration.
Co-reporter:Gary F. Moore, Michael Hambourger, Gerdenis Kodis, Weston Michl, Devens Gust, Thomas A. Moore, and Ana L. Moore
The Journal of Physical Chemistry B 2010 Volume 114(Issue 45) pp:14450-14457
Publication Date(Web):May 17, 2010
DOI:10.1021/jp101592m
The conversion of tyrosine to the corresponding tyrosyl radical in photosytem II (PSII) is an example of proton-coupled electron transfer. Although the tyrosine moiety (TyrZ) is known to function as a redox mediator between the photo-oxidized primary donor (P680•+) and the Mn-containing oxygen-evolving complex, the protonation states involved in the course of the reaction remain an active area of investigation. Herein, we report on the optical, structural, and electrochemical properties of tyrosine−histidine constructs, which model the function of their naturally occurring counterparts in PSII. Electrochemical studies show that the phenoxyl/phenol couple of the model is chemically reversible and thermodynamically capable of water oxidation. Studies under acidic and basic conditions provide clear evidence that an ionizable proton controls the electrochemical potential of the tyrosine−histidine mimic and that an exogenous base or acid can be used to generate a low-potential or high-potential mediator, respectively. The phenoxyl/phenoxide couple associated with the low-potential mediator is thermodynamically incapable of water oxidation, whereas the relay associated with the high-potential mediator is thermodynamically incapable of reducing an attached photoexcited porphyrin. These studies provide insight regarding the mechanistic role of the tyrosine−histidine complex in water oxidation and strategies for making use of hydrogen bonds to affect the coupling between proton and electron transfer in artificial photosynthetic systems.
Co-reporter:Michael Hambourger, Gary F. Moore, David M. Kramer, Devens Gust, Ana L. Moore and Thomas A. Moore
Chemical Society Reviews 2009 vol. 38(Issue 1) pp:25-35
Publication Date(Web):04 Nov 2008
DOI:10.1039/B800582F
Sunlight is the ultimate energy source for the vast majority of life on Earth, and organisms have evolved elegant machinery for energy capture and utilization. Solar energy, whether converted to wind, rain, biomass or fossil fuels, is also the primary energy source for human-engineered energy transduction systems. This tutorial review draws parallels between biological and technological energy systems. Aspects of biology that might be advantageously incorporated into emerging technologies are highlighted, as well as ways in which technology might improve upon the principles found in biological systems. Emphasis is placed upon artificial photosynthesis, as well as the use of protonmotive force in biology.
Co-reporter:Yuichi Terazono, Gerdenis Kodis, Paul A. Liddell, Vikas Garg, Thomas A. Moore, Ana L. Moore and Devens Gust
The Journal of Physical Chemistry B 2009 Volume 113(Issue 20) pp:7147-7155
Publication Date(Web):April 27, 2009
DOI:10.1021/jp900835s
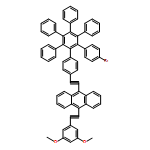
In order to ensure efficient utilization of the solar spectrum, photosynthetic organisms use a variety of antenna chromophores to absorb light and transfer excitation to a reaction center, where photoinduced charge separation occurs. Reported here is a synthetic molecular heptad that features two bis(phenylethynyl)anthracene and two borondipyrromethene antennas linked to a hexaphenylbenzene core that also bears two zinc porphyrins. A fullerene electron acceptor self-assembles to both porhyrins via dative bonds. Excitation energy is transferred very efficiently from all four antennas to the porphyrins. Singlet−singlet energy transfer occurs both directly and by a stepwise funnel-like pathway wherein excitation moves down a thermodynamic gradient. The porphyrin excited states donate an electron to the fullerene with a time constant of 3 ps to generate a charge-separated state with a lifetime of 230 ps. The overall quantum yield is close to unity. In the absence of the fullerene, the porphyrin excited singlet state donates an electron to a borondipyrromethene on a slower time scale. This molecule demonstrates that by incorporating antennas, it is possible for a molecular system to harvest efficiently light throughout the visible from ultraviolet wavelengths out to ∼650 nm.
Co-reporter:Joakim Andréasson Dr.;StephenD. Straight Dr.;ThomasA. Moore Dr.;AnaL. Moore Dr. Dr.
Chemistry - A European Journal 2009 Volume 15( Issue 16) pp:3936-3939
Publication Date(Web):
DOI:10.1002/chem.200900043
Co-reporter:Paul A. Liddell, Miguel Gervaldo, James W. Bridgewater, Amy E. Keirstead, Su Lin, Thomas A. Moore, Ana L. Moore and Devens Gust
Chemistry of Materials 2008 Volume 20(Issue 1) pp:135
Publication Date(Web):December 6, 2007
DOI:10.1021/cm7022955
The monomer 5-(4-aminophenyl)-10,20-bis(2,4,6-trimethylphenyl)porphyrin was synthesized and found to electropolymerize on platinum, indium tin oxide, and other electrodes to form a clear, semiconducting film with strong absorption in the visible spectral region. The linear, hole-conducting polymer has a unique structure, with porphyrin units linked to one another through the 5-(4-aminophenyl) nitrogen atom and the carbon atom at the 15-position on the macrocyclic ring. The porphyrin macrocyclic ring is thus an integral part of the linear polymer backbone. The oxidation potential of the film is 0.85 V and the reduction potential is −1.12 V vs SCE. The absorption spectrum of the film resembles that of a monomeric model porphyrin, but with significant peak broadening. Streak camera studies of the fluorescence of the polymer yield a lifetime of 15 ps, indicating strong quenching of the porphyrin first excited singlet state relative to that of the monomer. The properties of the polymer suggest that it may be useful in sensors, catalysts, and solar energy conversion devices.
Co-reporter:S. D. Straight;P. A. Liddell;Y. Terazono;A. L. Moore;T. A. Moore;D. Gust
Advanced Functional Materials 2007 Volume 17(Issue 5) pp:777-785
Publication Date(Web):29 JAN 2007
DOI:10.1002/adfm.200600802
A molecular triad consisting of a porphyrin linked to two photochromes, a fulgimide, and a dithienylethene, is synthesized and studied. When both photochromes are in their visible-light-absorbing forms, excitation of the fulgimide at 470 nm initiates a two-step singlet energy-transfer relay wherein excitation migrates first to the porphyrin and then to the dithienylethene. Photoisomerization of the dithienylethene to the open form using visible light prevents the second step, and excitation ultimately resides on the porphyrin, which fluoresces. Photoisomerization of the fulgimide eliminates significant absorption by the molecule at 470 nm, and consequently porphyrin excitation by energy transfer. Photoisomerization of each photochrome may be preferentially achieved, allowing access to all four isomeric states of the molecule. These states correspond to the outputs of logic gates, allowing solutions of the triad to perform either NOT-OR (NOR) or exclusive OR (XOR) functions using only optical inputs and outputs.
Co-reporter:Gerdenis Kodis;Yuichi Terazono;Paul A. Liddell;Miguel Gervaldo;Vikas Garg;Thomas A. Moore;Ana L. Moore
Photochemistry and Photobiology 2007 Volume 83(Issue 2) pp:464-469
Publication Date(Web):27 FEB 2007
DOI:10.1562/2006-12-05-RC-1098
A hexaphenylbenzene-based zinc porphyrin dyad forms a 1:1 complex with a fullerene bearing two pyridyl groups via coordination of the pyridyl nitrogens with the zinc atoms. The fullerene is symmetrically located between the two zinc porphyrins. The binding constant for the complex is 7.3 × 104 M−1 in 1,2-difluorobenzene. Photoinduced electron transfer from a porphyrin first excited singlet state to the fullerene occurs with a time constant of 3 ps, and the resulting charge-separated state has a lifetime of 230 ps. This self-assembled construct should form a basis for the construction of more elaborate model photosynthetic antenna-reaction center systems.
Co-reporter:Michael Hambourger, Paul A. Liddell, Devens Gust, Ana L. Moore and Thomas A. Moore
Photochemical & Photobiological Sciences 2007 vol. 6(Issue 4) pp:431-437
Publication Date(Web):12 Jan 2007
DOI:10.1039/B616444G
A hybrid photoelectrochemical biofuel cell employing the photoanode architecture of a dye-sensitized solar cell has been assembled. A porphyrin dye sensitizes a TiO2 semiconductor over the visible range to beyond 650 nm. Photoinduced charge separation at the dye–TiO2 interface results in electron migration to a cathode, and the holes generated on surface bound dyes oxidize soluble electron mediators. The increased [Ox]:[Red] ratio of the mediator drives the solution-based enzymatic oxidation of appropriate substrates. In this report we investigate how the accumulation of anodic and cathodic products limits cell performance. The NAD+/NADH and benzoquinone/hydroquinone redox couples were studied as sacrificial electron donors in the absence of appropriate enzymes or substrates. Comparatively poor cell performance was observed using the benzoquinone/hydroquinone couple. This effect is explained in terms of rapid charge recombination by electron donation from the electrode to benzoquinone in solution, as compared to much less recombination with NAD+. With the NAD+/NADH couple the cell performance is relatively independent of the redox poise of the anode solution, but limited by accumulation of reduction products in the cathodic compartment. Using the NAD+/NADH couple, the photochemical reforming of ethanol to hydrogen was demonstrated under conditions where the process would be endergonic in the dark.
Co-reporter:Joakim Andréasson Dr.;Stephen D. Straight;Subhajit Byopadhyay Dr.;Reginald H. Mitchell Dr.;Thomas A. Moore Dr.;Ana L. Moore Dr. Dr.
Angewandte Chemie 2007 Volume 119(Issue 6) pp:
Publication Date(Web):20 DEC 2006
DOI:10.1002/ange.200603856
Zwei in Eins: Ein an zwei photochrome Einheiten gebundenes Porphyrin fungiert als digitaler 2:1-Multiplexer (MUX). Wärme und rotes Licht sind die Eingangsdaten (ein 1 und ein 2), und ein dritter, schaltbarer Eingang (grünes Licht, sel) entscheidet, ob als Ausgang (Porphyrinfluoreszenz) der Zustand von „ein 1“ oder von „ein 2“ angezeigt wird. Jede photochrome Einheit lässt sich unabhängig so isomerisieren, dass sie die Porphyrinfluoreszenz löscht.
Co-reporter:Joakim Andréasson Dr.;Stephen D. Straight;Subhajit Byopadhyay Dr.;Reginald H. Mitchell Dr.;Thomas A. Moore Dr.;Ana L. Moore Dr. Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 6) pp:
Publication Date(Web):20 DEC 2006
DOI:10.1002/anie.200603856
Two into one: A porphyrin linked to two photochromic moieties performs as a 2:1 digital multiplexer (MUX). It takes heat and red light as the two inputs (in 1 and in 2), and a third switchable input (green light, sel) selects whether the output (porphyrin fluorescence) reports the state of in 1 or in 2. Each photochromic moiety may be independently photoisomerized to isomers that quench the porphyrin fluorescence.
Co-reporter:Rudi Berera, Gary F. Moore, Ivo H. M. van Stokkum, Gerdenis Kodis, Paul A. Liddell, Miguel Gervaldo, Rienk van Grondelle, John T. M. Kennis, Devens Gust, Thomas A. Moore and Ana L. Moore
Photochemical & Photobiological Sciences 2006 vol. 5(Issue 12) pp:1142-1149
Publication Date(Web):14 Nov 2006
DOI:10.1039/B613971J
We have designed and synthesized a molecular dyad comprising a carotenoid pigment linked to a fullerene derivative (C–C60) in which the carotenoid acts both as an antenna for the fullerene and as an electron transfer partner. Ultrafast transient absorption spectroscopy was carried out on the dyad in order to investigate energy transfer and charge separation pathways and efficiencies upon excitation of the carotenoid moiety. When the dyad is dissolved in hexane energy transfer from the carotenoid S2 state to the fullerene takes place on an ultrafast (sub 100 fs) timescale and no intramolecular electron transfer was detected. When the dyad is dissolved in toluene, the excited carotenoid decays from its excited states both by transferring energy to the fullerene and by forming a charge-separated C˙+–C60˙−. The charge-separated state is also formed from the excited fullerene following energy transfer from the carotenoid. These pathways lead to charge separation on the subpicosecond time scale (possibly from the S2 state and the vibrationally excited S1 state of the carotenoid), on the ps time scale (5.5 ps) from the relaxed S1 state of the carotenoid, and from the excited state of C60 in 23.5 ps. The charge-separated state lives for 1.3 ns and recombines to populate both the low-lying carotenoid triplet state and the dyad ground state.
Co-reporter:Joakim Andréasson, Yuichi Terazono, Bo Albinsson, Thomas A. Moore, Ana L. Moore,Devens Gust
Angewandte Chemie International Edition 2005 44(46) pp:7591-7594
Publication Date(Web):
DOI:10.1002/anie.200502420
Co-reporter:Joakim Andréasson Dr.;Yuichi Terazono Dr.;Bo Albinsson Dr.;Thomas A. Moore Dr.;Ana L. Moore Dr. Dr.
Angewandte Chemie 2005 Volume 117(Issue 46) pp:
Publication Date(Web):27 OCT 2005
DOI:10.1002/ange.200502420
Ein photochromer molekularer Schalter kann als Boolesches AND-Gate wirken. Als Eingabe dienen UV-Licht, das zur Photoisomerisierung eines Dihydroindolizins in die offene, dipolare Form führt, und ein elektrisches Feld, das dieses Isomer in Lösung ausrichtet (siehe Schema). Zum Lesen der Ausgabe wird der elektrische Lineardichroismus des offenen Isomers bestimmt. Sichtbares Licht stellt die Ausgangssituation wieder her.
Co-reporter:Rodrigo E. Palacios Dr.;Gerdenis Kodis Dr.;Stephanie L. Gould Dr.;Linda de la Garza Dr.;Alicia Brune Dr. ;Thomas A. Moore ;Ana L. Moore
ChemPhysChem 2005 Volume 6(Issue 11) pp:
Publication Date(Web):7 NOV 2005
DOI:10.1002/cphc.200500177
An artificial photosynthetic reaction center consisting of a carotenoid (C), a dimesitylporphyrin (P), and a bis(heptafluoropropyl)porphyrin (PF), C-P-PF , and the related triad in which the central porphyrin has been metalated to give C-PZn-PFhave been synthesized and characterized by transient spectroscopy. These triads are models for amphipathic triads having a carboxylate group attached to the PFmoiety; they are designed to carry out redox processes across lipid bilayers. Triad C-P-PFundergoes rapid singlet–singlet energy transfer between the porphyrin moieties, so that their excited states are in equilibrium. In benzonitrile, photoinduced electron transfer from the first excited singlet state of P and hole transfer from the first excited singlet state of PFyield the initial charge-separated state C-P.+-PF.−. Subsequent hole transfer to the carotenoid moiety generates the final charge-separated state C.+-P-PF.−, which has a lifetime of 1.1 μs and is formed with a quantum yield of 0.24. In triad C-PZn-PFenergy transfer from the PZnexcited singlet to the PFmoiety yields C-PZn-1PF . A series of electron-transfer reactions analogous to those observed in C-P-PFgenerates C.+-PZn-PF.−, which has a lifetime of 750 ns and is formed with a quantum yield of 0.25. Flash photolysis experiments in liposomes containing an amphipathic version of C-PZn-PFdemonstrate that the added driving force for photoinduced electron transfer in the metalated triad is useful for promoting electron transfer in the low-dielectric environment of artificial biological membranes. In argon-saturated toluene solutions of C-P-PFand C-PZn-PF , charge separation is not observed and a considerable yield of triplet species is generated upon excitation of the porphyrin moieties. In both triads triplet energy localized in the PFmoiety is channeled to the carotenoid chromophore by a triplet energy-transfer relay mechanism. Certain photophysical characteristics of these triads, including the sequential electron transfer and the triplet energy-transfer relay mechanism, are reminiscent of those observed in natural reaction centers of photosynthetic bacteria.
Co-reporter:Paul A. Liddell, Gerdenis Kodis, Darius Kuciauskas, Joakim Andréasson, Ana L. Moore, Thomas A. Moore and Devens Gust
Physical Chemistry Chemical Physics 2004 vol. 6(Issue 24) pp:5509-5515
Publication Date(Web):03 Nov 2004
DOI:10.1039/B412326C
Two triad molecules consisting of either two zinc, or two free-base porphyrins symmetrically joined to a fullerene via phenyleneethynylene-containing linkages have been synthesized, and their photochemistry investigated. In the zinc form of the triad, PZn–C60–PZn, excitation of a zinc porphyrin in 2-methyltetrahydrofuran solution is followed by photoinduced electron transfer to the fullerene with a time constant of 20 ps. The resulting PZn˙+–C60˙−–PZn charge-separated state is formed with a quantum yield of 98% and has a lifetime of 820 ps. The first excited singlet state of the free-base analog gives the P2H˙+–C60˙−–P2H charge-separated state with a time constant of 200 ps and a yield of 98%. The charge-separated state decays with a lifetime of 2.8 ns. The difference in the rates of photoinduced electron transfer is consistent with reaction in the normal region of the Marcus–Hush relationship of transfer rate and driving force, and charge recombination is consistent with Marcus–Hush inverted behavior. The presence of the two porphyrin electron donors in these triads enhances the absorption cross section for light collection, and the molecular framework employed could be used to prepare molecules with enhanced energy conversion or optoelectronic properties.
Co-reporter:Gerdenis Kodis;Paul A. Liddell;Ana L. Moore;Thomas A. Moore
Journal of Physical Organic Chemistry 2004 Volume 17(Issue 9) pp:724-734
Publication Date(Web):30 JUL 2004
DOI:10.1002/poc.787
A new photosynthetic reaction center mimic consisting of a porphyrin (P) linked to both a fullerene electron acceptor (C60) and a carotenoid secondary electron donor (C) was synthesized and studied in 2-methyltetrahydrofuran using transient spectroscopic methods. Excitation of the porphyrin is followed by photoinduced electron transfer to the fullerene (τ = 32 ps) to yield C–P·+–C60·−. Electron transfer from the carotene to the porphyrin radical cation (τ = 125 ps) gives a final C·+–P–C60·− state with an overall yield of 0.95. This state decays to give the carotenoid triplet state with a time constant of 57 ns. The molecular triad is highly soluble in organic solvents and readily synthesized. These qualities make the molecule a useful artificial photosynthetic reaction center for a variety of spectroscopic and photochemical investigations. Copyright © 2004 John Wiley & Sons, Ltd.
Co-reporter:Gerdenis Kodis, Paul A. Liddell, Linda de la Garza, Ana L. Moore, Thomas A. Moore and Devens Gust
Journal of Materials Chemistry A 2002 vol. 12(Issue 7) pp:2100-2108
Publication Date(Web):22 Apr 2002
DOI:10.1039/B201039A
Two molecular triads consisting of a porphyrin (P) covalently linked to a fullerene electron acceptor (C60) and a π-extended tetrathiafulvalene electron donor (TTF) have been synthesized. Time resolved spectroscopic investigations of the triad featuring a free base porphyrin moiety (TTF–P2H–C60) show that in 2-methyltetrahydrofuran solution, excitation of the porphyrin leads to formation of a TTF–P2H˙+–C60˙− charge-separated state in 25 ps. Electron transfer from the TTF generates a final TTF˙+–P2H–C60˙−state with an overall yield of 0.87. This species decays to the ground state in 1.07 µs. Similar experiments on the zinc analog, TTF–PZn–C60, show formation of TTF–PZn˙+–C60˙−
in 1.5 ps, followed by generation of TTF˙+–PZn–C60˙− with a yield of 0.09. This charge-separated state also decays to the ground state in 1.07 µs. Comparison of these results with those for previously reported triads with different donor moieties reveals differences in electron transfer rate constants that can be qualitatively understood in the framework of the Marcus–Hush electron transfer formalism.
Co-reporter:Ira M. Bennett;Hebe M. Vanegas Farfano;Federica Bogani;Alex Primak;Paul A. Liddell;Luis Otero;Leonides Sereno;Juana J. Silber;Ana L. Moore;Thomas A. Moore
Nature 2002 420(6914) pp:398-401
Publication Date(Web):2002-11-28
DOI:10.1038/nature01209
Transport of calcium ions across membranes and against a thermodynamic gradient is essential to many biological processes, including muscle contraction, the citric acid cycle, glycogen metabolism, release of neurotransmitters, vision, biological signal transduction and immune response. Synthetic systems that transport metal ions across lipid or liquid membranes are well known1, 2, 3, 4, 5, 6, and in some cases light has been used to facilitate transport7. Typically, a carrier molecule located in a symmetric membrane binds the ion from aqueous solution on one side and releases it on the other. The thermodynamic driving force is provided by an ion concentration difference between the two aqueous solutions, coupling to such a gradient in an auxiliary species, or photomodulation of the carrier by an asymmetric photon flux7. Here we report a different approach, in which active transport is driven not by concentration gradients, but by light-induced electron transfer in a photoactive molecule that is asymmetrically disposed across a lipid bilayer. The system comprises a synthetic, light-driven transmembrane Ca2+ pump based on a redox-sensitive, lipophilic Ca2+-binding shuttle molecule whose function is powered by an intramembrane artificial photosynthetic reaction centre. The resulting structure transports calcium ions across the bilayer of a liposome to develop both a calcium ion concentration gradient and a membrane potential, expanding Mitchell's concept of a redox loop mechanism for protons8 to include divalent cations. Although the quantum yield is relatively low (~1 per cent), the Ca2+ electrochemical potential developed is significant.
Co-reporter:Darius Kuciauskas;Paul A. Liddell;Jeffrey L. Bahr;Thomas A. Moore;Ana L. Moore
Photochemistry and Photobiology 2000 Volume 72(Issue 5) pp:598-611
Publication Date(Web):1 MAY 2007
DOI:10.1562/0031-8655(2000)0720598DFAECE2.0.CO2
Tuning thermodynamic driving force and electronic coupling through structural modifications of a carotene (C) porphyrin (P) fullerene (C60) molecular triad has permitted control of five electron and energy transfer rate constants and two excited state lifetimes in order to prepare a high-energy charge-separated state by photoinduced electron transfer with a quantum yield of essentially unity (≥96%). Excitation of the porphyrin moiety of C–P–C60 is followed by a combination of photoinduced electron transfer to give C–P·+–C60·− and singlet–singlet energy transfer to yield C–P–1C60. The fullerene excited state accepts an electron from the porphyrin to also generate C–P·+–C60·−. Overall, this initial state is formed with a quantum yield of 0.97. Charge shift from the carotenoid to yield C·+–P–C60·− is at least 60 times faster than recombination of C–P·+–C60·−, leading to the overall quantum yield near unity for the final state. Formation of a similar charge-separated species from the zinc analog of the triad with a yield of 40% is also observed. Charge recombination of C·+–P–C60·− in 2-methyltetrahydrofuran yields the carotenoid triplet state, rather than the ground state. Comparison of the results for this triad with those for related triads with different structural features provides information concerning the effects of driving force and electronic coupling on each of the electron transfer steps.
Co-reporter:Matthieu Koepf, Jesse J. Bergkamp, Anne-Lucie Teillout, Manuel J. Llansola-Portoles, Gerdenis Kodis, Ana L. Moore, Devens Gust and Thomas A. Moore
Dalton Transactions 2017 - vol. 46(Issue 13) pp:NaN4208-4208
Publication Date(Web):2017/02/21
DOI:10.1039/C6DT04647A
The association of different metals in stable, well-defined molecular assemblies remains a great challenge of supramolecular chemistry. In such constructs, the emergence of synergism, or cooperative effects between the different metal centers is particularly intriguing. These effects can lead to uncommon reactivity or remarkable physico-chemical properties that are not otherwise achievable. For example, the association of alkaline or alkaline-earth cations and transition metals is pivotal for the activity of several biomolecules and human-made catalysts that carry out fundamental redox transformations (water oxidation, nitrogen reduction, water–gas shift reaction, etc.). In many cases the precise nature of the interactions between the alkaline-earth cations and the redox-active transition metals remains elusive due to the difficulty of building stable molecular heterometallic assemblies that associate transition metals and alkaline or alkaline-earth cations in a controlled way. In this work we present the rational design of porphyrin-based ligands possessing a second binding site for alkaline-earth cations above the porphyrin macrocycle primary complexation site. We demonstrate that by using a combination of crown ether and carboxylic acid substituents suitably positioned on the periphery of the porphyrin, bitopic ligands can be obtained. The binding of calcium, a typical alkaline-earth cation, by the newly prepared ligands has been studied in detail and we show that a moderately large binding constant can be achieved in protic media using ligands that possess some degree of structural flexibility. The formation of Zn–Ca assemblies discussed in this work is viewed as a stepping stone towards the assembly of well defined molecular transition metal-alkaline earth bimetallic centers using a versatile organic scaffold.
Co-reporter:Michael Hambourger, Gary F. Moore, David M. Kramer, Devens Gust, Ana L. Moore and Thomas A. Moore
Chemical Society Reviews 2009 - vol. 38(Issue 1) pp:NaN35-35
Publication Date(Web):2008/11/04
DOI:10.1039/B800582F
Sunlight is the ultimate energy source for the vast majority of life on Earth, and organisms have evolved elegant machinery for energy capture and utilization. Solar energy, whether converted to wind, rain, biomass or fossil fuels, is also the primary energy source for human-engineered energy transduction systems. This tutorial review draws parallels between biological and technological energy systems. Aspects of biology that might be advantageously incorporated into emerging technologies are highlighted, as well as ways in which technology might improve upon the principles found in biological systems. Emphasis is placed upon artificial photosynthesis, as well as the use of protonmotive force in biology.
Co-reporter:A. Antoniuk-Pablant, Y. Terazono, B. J. Brennan, B. D. Sherman, J. D. Megiatto, G. W. Brudvig, A. L. Moore, T. A. Moore and D. Gust
Journal of Materials Chemistry A 2016 - vol. 4(Issue 8) pp:NaN2985-2985
Publication Date(Web):2015/10/16
DOI:10.1039/C5TA07226C
β-Cyanoporphyrins have high positive potentials for oxidation and absorb light at longer wavelengths than most porphyrins, making them potential candidates for sensitizers in photoelectrosynthetic cells for water oxidation. In order to begin to evaluate this potential, two Zn(II) tetra-β-cyanoporphyrins have been synthesized and evaluated as sensitizers in dye sensitized solar cells using I−/I3− as the redox mediator. To prepare such specialized β-cyanoporphyrins, a new synthetic method has been developed. This approach involves reaction of Zn(CN)2 with β-brominated zinc porphyrins in the presence of tris-(dibenzylideneacetone)dipalladium. The tetra-cyanation reaction is complete under milder conditions as compared to those usually employed in previous methods and gives improved yields of up to ∼50%. The procedure allows for the cyanation of porphyrins with relatively sensitive functional groups. Examples of its application to a range of substituted tetra-arylporphyrins are reported, and the absorption and electrochemical properties of the compounds prepared are given. The results from using two of the molecules as sensitizers in dye sensitized solar cells are presented. It was found that the porphyrins produced no photocurrents in nanoparticulate TiO2-based cells, but both molecules produced photocurrents in SnO2-based cells, and are potential candidates for sensitizers in photoelectrosynthetic cells for water oxidation.
Co-reporter:Devens Gust, Joakim Andréasson, Uwe Pischel, Thomas A. Moore and Ana L. Moore
Chemical Communications 2012 - vol. 48(Issue 14) pp:NaN1957-1957
Publication Date(Web):2011/12/05
DOI:10.1039/C1CC15329C
Photochromes are chromophores that are reversibly isomerized between two metastable forms using light, or light and heat. When photochromes are covalently linked to other chromophores, they can act as molecular photonic analogues of electronic transistors. As bistable switches, they can be incorporated into the design of molecules capable of binary arithmetic and both combinatorial and sequential digital logic operations. Small ensembles of such molecules can perform analogue signal modulation similar to that carried out by transistor amplifiers. Examples of molecules that perform multiple logic functions, act as control elements for fluorescent reporters, and mimic natural photoregulatory functions are presented.
Co-reporter:Bradley J. Brennan, Michael J. Kenney, Paul A. Liddell, Brian R. Cherry, Jian Li, Ana L. Moore, Thomas A. Moore and Devens Gust
Chemical Communications 2011 - vol. 47(Issue 36) pp:NaN10036-10036
Publication Date(Web):2011/08/09
DOI:10.1039/C1CC13596A
A method for radical coupling of porphyrins using copper(II) salts as one-electron oxidants was developed. A Zn(II)-porphyrin bearing an aminophenyl group yielded porphyrin oligomers, and two tri-arylporphyrins were oxidized to form doubly and triply linked dimers. Bromination of doubly linked dimers gave macrocycles with twisted skeletons.
Co-reporter:Bradley J. Brennan, Manuel J. Llansola Portolés, Paul A. Liddell, Thomas A. Moore, Ana L. Moore and Devens Gust
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 39) pp:NaN16614-16614
Publication Date(Web):2013/07/25
DOI:10.1039/C3CP52156G
A tetra-arylporphyrin dye was functionalized with three different anchoring groups used to attach molecules to metal oxide surfaces. The physical, photophysical and electrochemical properties of the derivatized porphyrins were studied, and the dyes were then linked to mesoporous TiO2. The anchoring groups were β-vinyl groups bearing either a carboxylate, a phosphonate or a siloxy moiety. The siloxy linkages were made by treatment of the metal oxide with a silatrane derivative of the porphyrin. The surface binding and lability of the anchored molecules were studied, and dye performance was compared in a dye-sensitized solar cell (DSSC). Transient absorption spectroscopy was used to study charge recombination processes. At comparable surface concentration, the porphyrin showed comparable performance in the DSSC, regardless of the linker. However, the total surface coverage achievable with the carboxylate was about twice that obtainable with the other two linkers, and this led to higher current densities for the carboxylate DSSC. On the other hand, the carboxylate-linked dyes were readily leached from the metal oxide surface under alkaline conditions. The phosphonates were considerably less labile, and the siloxy-linked porphyrins were most resistant to leaching from the surface. The use of silatrane proved to be a practical and convenient way to introduce the siloxy linkages, which can confer greatly increased stability on dye-sensitized electrodes with photoelectrochemical performance comparable to that of the other linkers.
Co-reporter:Robert A. Schmitz, Paul A. Liddell, Gerdenis Kodis, Michael J. Kenney, Bradley J. Brennan, Nolan V. Oster, Thomas A. Moore, Ana L. Moore and Devens Gust
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 33) pp:
Publication Date(Web):
DOI:10.1039/C4CP02105C
Co-reporter:Brian L. Watson, Benjamin D. Sherman, Ana L. Moore, Thomas A. Moore and Devens Gust
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 24) pp:NaN15796-15796
Publication Date(Web):2015/05/18
DOI:10.1039/C5CP00860C
A new sensitizer motif for dye sensitized solar cells (DSSC) has been developed. A heteroaromatic moiety containing a pyrazine ring links two porphyrin chromophores to the metal oxide surface via two carboxylic acid attachment groups. A test DSSC sensitized with the new molecule was 3.5 times more efficient than a similar cell sensitized by a single porphyrin model compound. The open circuit photovoltage was increased by a modest factor of 1.3, but the photocurrent increased by a factor of 2.7. Most of the increase is attributed to a reduced rate of charge recombination of the charge separated state formed by photoinduced electron transfer from the excited sensitizer to the TiO2, although some of the difference is due to increased light absorption resulting from more dye on the photoanode. Increased light absorption due to the pyrazine-containing group may also play a role. The design illustrated here could also be used to link complementary sensitizers or antenna moieties in order to increase spectral coverage.
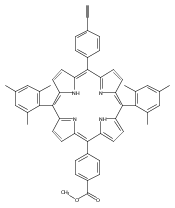
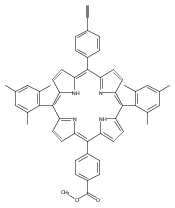
.jpg)


![1H-Pyrrole, 2,2'-[[4-(1,1-dimethylethyl)phenyl]methylene]bis-](/data/chemimg/909900/167482-98-6.png)
![1H-Pyrrole, 2,2'-[[4-(1,1-dimethylethyl)phenyl]methylene]bis-](/data/chemimg/909900/167482-98-6_b.png)





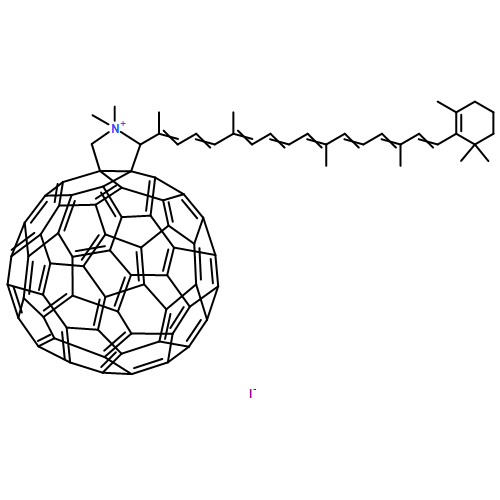
![Thiophene, 5-(4-ethynylphenyl)-3-[3,3,4,4,5,5-hexafluoro-2-[5-(4-methoxyphenyl)-2-methyl-3-thienyl]-1-cyclopenten-1-yl]-2-methyl-](/data/chemimg/126700/444689-09-2.png)
![Thiophene, 5-(4-ethynylphenyl)-3-[3,3,4,4,5,5-hexafluoro-2-[5-(4-methoxyphenyl)-2-methyl-3-thienyl]-1-cyclopenten-1-yl]-2-methyl-](/data/chemimg/126700/444689-09-2_b.png)



![Silane, [(4-bromophenyl)ethynyl]tris(1-methylethyl)-](http://img.cochemist.com/ccimg/271300/271242-20-7.png)
![Silane, [(4-bromophenyl)ethynyl]tris(1-methylethyl)-](http://img.cochemist.com/ccimg/271300/271242-20-7_b.png)












![DICYCLOPENTA[CD,FG]PYRENE](http://img.cochemist.com/ccimg/173700/173678-72-3.png)
![DICYCLOPENTA[CD,FG]PYRENE](http://img.cochemist.com/ccimg/173700/173678-72-3_b.png)





![2,2'-[(4-nitrophenyl)methylene]bis-1H-Pyrrole](http://img.cochemist.com/ccimg/143900/143859-77-2.png)
![2,2'-[(4-nitrophenyl)methylene]bis-1H-Pyrrole](http://img.cochemist.com/ccimg/143900/143859-77-2_b.png)


![Cyclopent[hi]acephenanthrylene](http://img.cochemist.com/ccimg/115000/114959-37-4.png)
![Cyclopent[hi]acephenanthrylene](http://img.cochemist.com/ccimg/115000/114959-37-4_b.png)
![Dicyclopenta[cd,jk]pyrene](http://img.cochemist.com/ccimg/98800/98791-43-6.png)
![Dicyclopenta[cd,jk]pyrene](http://img.cochemist.com/ccimg/98800/98791-43-6_b.png)
![Dicyclopenta[cd,mn]pyrene](http://img.cochemist.com/ccimg/97000/96915-04-7.png)
![Dicyclopenta[cd,mn]pyrene](http://img.cochemist.com/ccimg/97000/96915-04-7_b.png)
![Dicyclopenta[cd,lm]perylene](http://img.cochemist.com/ccimg/80900/80879-80-7.png)
![Dicyclopenta[cd,lm]perylene](http://img.cochemist.com/ccimg/80900/80879-80-7_b.png)
![Phosphonium, [(4-iodophenyl)methyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/61200/61130-13-0.png)
![Phosphonium, [(4-iodophenyl)methyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/61200/61130-13-0_b.png)
![11-oxo-2,3,5,6,7,11-Hexahydro-1H-pyrano[2,3-f]pyrido[3,2,1-ij]quinoline-10-carboxylic acid](/data/chemimg/86700/55804-65-4.png)
![11-oxo-2,3,5,6,7,11-Hexahydro-1H-pyrano[2,3-f]pyrido[3,2,1-ij]quinoline-10-carboxylic acid](/data/chemimg/86700/55804-65-4_b.png)
![Cyclopenta[cd]pyrene](http://img.cochemist.com/ccimg/27300/27208-37-3.png)
![Cyclopenta[cd]pyrene](http://img.cochemist.com/ccimg/27300/27208-37-3_b.png)


![Phenoxy, 4-[(2S)-2-amino-2-carboxyethyl]-](/data/chemimg/1149200/16978-66-8.png)
![Phenoxy, 4-[(2S)-2-amino-2-carboxyethyl]-](/data/chemimg/1149200/16978-66-8_b.png)
![Zincate(2-), [7,12-bis(1-hydroxyethyl)-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/472300/13939-11-2.png)
![Zincate(2-), [7,12-bis(1-hydroxyethyl)-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/472300/13939-11-2_b.png)



![dibenzo[ghi,mno]fluoranthene](http://img.cochemist.com/ccimg/5900/5821-51-2.png)
![dibenzo[ghi,mno]fluoranthene](http://img.cochemist.com/ccimg/5900/5821-51-2_b.png)









![Dibenz[a,j]anthracene](http://img.cochemist.com/ccimg/300/224-41-9.png)
![Dibenz[a,j]anthracene](http://img.cochemist.com/ccimg/300/224-41-9_b.png)
![Cyclopenta[hi]chrysene(8CI,9CI)](http://img.cochemist.com/ccimg/300/216-48-8.png)
![Cyclopenta[hi]chrysene(8CI,9CI)](http://img.cochemist.com/ccimg/300/216-48-8_b.png)


![Benzo[ghi]fluoranthene](http://img.cochemist.com/ccimg/300/203-12-3.png)
![Benzo[ghi]fluoranthene](http://img.cochemist.com/ccimg/300/203-12-3_b.png)




![dibenzo[fg,ij]pentaphene](http://img.cochemist.com/ccimg/200/197-69-3.png)
![dibenzo[fg,ij]pentaphene](http://img.cochemist.com/ccimg/200/197-69-3_b.png)

![cyclohepta[fg]acenaphthylene](http://img.cochemist.com/ccimg/200/194-32-1.png)
![cyclohepta[fg]acenaphthylene](http://img.cochemist.com/ccimg/200/194-32-1_b.png)
![Cyclopenta[cd]fluoranthene](http://img.cochemist.com/ccimg/200/193-54-4.png)
![Cyclopenta[cd]fluoranthene](http://img.cochemist.com/ccimg/200/193-54-4_b.png)
![Cyclopenta[cd]perylene](http://img.cochemist.com/ccimg/200/189-01-5.png)
![Cyclopenta[cd]perylene](http://img.cochemist.com/ccimg/200/189-01-5_b.png)
![Dinaphtho[2,1,8,7-defg:2',1',8',7'-opqr]pentacene](http://img.cochemist.com/ccimg/200/188-91-0.png)
![Dinaphtho[2,1,8,7-defg:2',1',8',7'-opqr]pentacene](http://img.cochemist.com/ccimg/200/188-91-0_b.png)
![Cyclopent[fg]acenaphthylene](http://img.cochemist.com/ccimg/200/187-78-0.png)
![Cyclopent[fg]acenaphthylene](http://img.cochemist.com/ccimg/200/187-78-0_b.png)




![2,2'-[(2,3,4,5,6-pentafluorophenyl)methylene]bis-1H-Pyrrole](http://img.cochemist.com/ccimg/167500/167482-91-9.png)
![2,2'-[(2,3,4,5,6-pentafluorophenyl)methylene]bis-1H-Pyrrole](http://img.cochemist.com/ccimg/167500/167482-91-9_b.png)