Co-reporter:Ariana Gray Bé, Mary Alice Upshur, Pengfei Liu, Scot T. Martin, Franz M. Geiger, and Regan J. Thomson
ACS Central Science July 26, 2017 Volume 3(Issue 7) pp:715-715
Publication Date(Web):July 5, 2017
DOI:10.1021/acscentsci.7b00112
The formation of atmospheric cloud droplets due to secondary organic aerosol (SOA) particles is important for quantifying the Earth’s radiative balance under future, possibly warmer, climates, yet is only poorly understood. While cloud activation may be parametrized using the surface tension depression that coincides with surfactant partitioning to the gas–droplet interface, the extent to which cloud activation is influenced by both the chemical structure and reactivity of the individual molecules comprising this surfactant pool is largely unknown. We report herein considerable differences in the surface tension depression of aqueous pendant droplets that contain synthetically prepared ozonolysis products derived from α-pinene and β-caryophyllene, the most abundant of the monoterpenes and sesquiterpenes, respectively, that are emitted over the planet’s vast forest ecosystems. Oxidation products derived from β-caryophyllene were found to exhibit significantly higher surface activity than those prepared from α-pinene, with the critical supersaturation required for cloud droplet activation reduced by 50% for β-caryophyllene aldehyde at 1 mM. These considerable reductions in the critical supersaturation were found to coincide with free energies of adsorption that exceed ∼25 kJ/mol, or just one hydrogen bond equivalent, depending on the ammonium sulfate and oxidation product concentration in the solution. Additional experiments showed that aldehyde-containing oxidation products exist in equilibrium with hydrated forms in aqueous solution, which may modulate their bulk solubility and surface activity. Equilibration time scales on the order of 10–5 to 10–4 s calculated for micrometer-sized aerosol particles indicate instantaneous surface tension depression in the activation processes leading to cloud formation in the atmosphere. Our findings highlight the underlying importance of molecular structure and reactivity when considering cloud condensation activity in the presence of SOA particles.
Co-reporter:Merve Doğangün, Mimi N. Hang, Jo Machesky, Alicia C. McGeachy, Naomi Dalchand, Robert J. Hamers, and Franz M. Geiger
The Journal of Physical Chemistry C December 14, 2017 Volume 121(Issue 49) pp:27473-27473
Publication Date(Web):November 10, 2017
DOI:10.1021/acs.jpcc.7b09187
An experimental investigation of how electrostatics and ion dissolution impact the interaction between nanosheets of lithium intercalation compounds and supported lipid bilayers has revealed evidence for considerable metal cation concentrations in the nanosheets–bilayer (the “nano–bio interface”) gap. Specifically, elevated concentrations of aqueous metal ions in the 1 mg/L concentration regime produce vibrational sum frequency generation signal intensity changes that are commensurate with the induction of compositional membrane asymmetry. This outcome is consistent with the notion that the induction of bilayer asymmetry by LiCoO2 nanosheets occurs through a noncontact mechanism that primarily involves the interaction of negatively charged lipids with dissolved ions concentrated within the electrical double layers present in the nano–bio interface gap. Our findings provide a possible avenue for redesign strategies that mitigate noncontact interactions between nanomaterials and biological interfaces, enabling the design of new energy storage materials with reduced environmental impacts.
Co-reporter:Alicia C. McGeachy, Laura L. Olenick, Julianne M. Troiano, Ronald S. Lankone, Eric S. Melby, Thomas R. Kuech, Eseohi Ehimiaghe, D. Howard Fairbrother, Joel A. PedersenFranz M. Geiger
The Journal of Physical Chemistry B 2017 Volume 121(Issue 6) pp:
Publication Date(Web):January 13, 2017
DOI:10.1021/acs.jpcb.6b10141
With production of carbon nanotubes surpassing billions of tons per annum, concern about their potential interactions with biological systems is growing. Herein, we utilize second harmonic generation spectroscopy, sum frequency generation spectroscopy, and quartz crystal microbalance with dissipation monitoring to probe the interactions between oxidized multiwalled carbon nanotubes (O-MWCNTs) and supported lipid bilayers composed of phospholipids with phosphatidylcholine head groups as the dominant component. We quantify O-MWCNT attachment to supported lipid bilayers under biogeochemically relevant conditions and discern that the interactions occur without disrupting the structural integrity of the lipid bilayers for the systems probed. The extent of O-MWCNT sorption was far below a monolayer even at 100 mM NaCl and was independent of the chemical composition of the supported lipid bilayer.
Co-reporter:Hilary M. Chase, Shunli Chen, Li Fu, Mary Alice Upshur, Benjamin Rudshteyn, Regan J. Thomson, Hong-Fei Wang, Victor S. Batista, Franz M. Geiger
Chemical Physics Letters 2017 Volume 683(Volume 683) pp:
Publication Date(Web):1 September 2017
DOI:10.1016/j.cplett.2017.01.015
•Orientations of small molecules are determined by experimental and theoretical SFG.•Orientations cannot be derived from experiment alone when modes are delocalized.•Theoretical SFG produces spectra that match experiment for all six compounds.Inferring molecular orientations from vibrational sum frequency generation (SFG) spectra is challenging in polarization combinations that result in low signal intensities, or when the local point group symmetry approximation fails. While combining experiments with density functional theory (DFT) could overcome this problem, the scope of the combined method has yet to be established. Here, we assess its feasibility of determining the distributions of molecular orientations for one monobasic ester, two epoxides and three alcohols at the vapor/fused silica interface. We find that molecular orientations of nonlocal vibrational modes cannot be determined using polarization-resolved SFG measurements alone.Download high-res image (109KB)Download full-size image
Co-reporter:Julianne M. Troiano, Thomas R. Kuech, Ariane M. Vartanian, Marco D. Torelli, Akash Sen, Lisa M. Jacob, Robert J. Hamers, Catherine J. Murphy, Joel A. Pedersen, and Franz M. Geiger
The Journal of Physical Chemistry C 2016 Volume 120(Issue 37) pp:20659-20667
Publication Date(Web):April 12, 2016
DOI:10.1021/acs.jpcc.6b01786
Second harmonic generation (SHG) is useful for studying the properties of interfaces, including the surfaces of nanoparticles and the interaction of nanoparticles with biologically relevant surfaces. Gold nanoparticles at the biological membrane represent a particularly interesting system to be probed by SHG spectroscopy given the rich electronic structure of gold nanoparticles and the charged nature of the nano-bio interface. Here we describe the interplay between the resonant and nonresonant components of the second harmonic response as 4 and 14 nm spherical gold nanoparticles (AuNPs) wrapped in the cationic polyelectrolyte poly(allylamine hydrochloride) (PAH) adsorb to negatively charged supported lipid bilayers. In contrast to the SHG response of 4 nm PAH-AuNPs, that we have shown previously to be dominated by resonance enhancement, the SHG response from the adsorption of the 14 nm PAH-AuNPs, with similar hydrodynamic diameters, to a 9:1 DOPC:DOTAP bilayer is dominated by the nonresonant, interfacial, potential-dependent component of the signal. We hypothesize that the difference in the SHG response is attributable to the differences in the number of PAH molecules associated with the particles and, therefore, differences in the number of positively charged ammonium groups associated with the 4 vs the 14 nm particles. For 14 nm PAH-AuNPs with larger hydrodynamic diameters, we determined two regimes in the adsorption behavior, one where the resonance enhancement from the gold core of the nanoparticle dominates the signal and a second where the nonresonant, interfacial, potential-dependent term dominates the signal. The results presented in this study provide insight into the interplay between resonant and nonresonant components of the second harmonic signal from the adsorption of charged AuNPs and are valuable for future studies with other functionalized particles and lipid systems by SHG.
Co-reporter:Junming Ho
The Journal of Physical Chemistry C 2016 Volume 120(Issue 23) pp:12578-12589
Publication Date(Web):May 17, 2016
DOI:10.1021/acs.jpcc.6b03158
A rotationally fluid state of α-pinene at fused silica/vapor interfaces is revealed by computational and experimental vibrational sum frequency generation (SFG) studies. We report the first assignment of the vibrational modes in the notoriously congested C–H stretching region of α-pinene and identify its bridge methylene group on the four-membered ring (“βCH2”) as the origin of its dominant spectral feature. We find that the spectra are perfused with Fermi resonances that need to be accounted for explicitly in the computation of vibrational spectra of strained hydrocarbons such as α-pinene. The preferred orientations of α-pinene are consistent with optimization of van der Waals contacts with the silica surface that results in a bimodal distribution of highly fluxional orientations in which the βCH2 group points “towards” or “away from” the surface. Classical molecular dynamics simulations further provide rotational diffusion constants of 49 ± 1 ps and 2580 ± 60 ps, which are attributed to two broad types of adsorption modes on silica. The reported findings are particularly relevant to the exposure of α-pinene to primary oxidants in heterogeneous catalytic pathways that exploit α-pinene as a sustainable feedstock for fine chemicals and polymers.
Co-reporter:Mary Alice Upshur, Hilary M. Chase, Benjamin F. Strick, Carlena J. Ebben, Li Fu, Hongfei Wang, Regan J. Thomson, and Franz M. Geiger
The Journal of Physical Chemistry A 2016 Volume 120(Issue 17) pp:2684-2690
Publication Date(Web):April 9, 2016
DOI:10.1021/acs.jpca.6b01995
This study aims to reliably assign the vibrational sum frequency generation (SFG) spectrum of α-pinene at the vapor/solid interface using a method involving deuteration of various methyl groups. The synthesis of five deuterated isotopologues of α-pinene is presented to determine the impact that removing contributions from methyl group C–H oscillators has on its SFG response. 0.6 cm–1 resolution SFG spectra of these isotopologues show varying degrees of differences in the C–H stretching region when compared to the SFG response of unlabeled α-pinene. The largest spectral changes were observed for the isotopologue containing a fully deuterated vinyl methyl group. Noticeable losses in signal intensities allow us to reliably assign the 2860 cm–1 peak to the vinyl methyl symmetric stretch. Furthermore, upon removing the vinyl methyl group entirely by synthesizing apopinene, the steric influence of the unlabeled C9H14 fragment on the SFG response of α-pinene SFG can be readily observed. The work presented here brings us one step closer to understanding the vibrational spectroscopy of α-pinene.
Co-reporter:Hilary M. Chase, Benjamin Rudshteyn, Brian T. Psciuk, Mary Alice Upshur, Benjamin F. Strick, Regan J. Thomson, Victor S. Batista, and Franz M. Geiger
The Journal of Physical Chemistry B 2016 Volume 120(Issue 8) pp:1919-1927
Publication Date(Web):November 12, 2015
DOI:10.1021/acs.jpcb.5b09769
We assess the capabilities of eight popular density functional theory (DFT) functionals, in combination with several basis sets, as applied to calculations of vibrational sum frequency generation (SFG) spectra of the atmospherically relevant isoprene oxidation product trans-β-isoprene epoxydiol (IEPOX) and one of its deuterated isotopologues at the fused silica/vapor interface. We use sum of squared differences (SSD) and total absolute error (TAE) calculations to estimate the performance of each functional/basis set combination in producing SFG spectra that match experimentally obtained spectra from trans-β-IEPOX and one of its isotopologues. Our joined SSD/TAE analysis shows that while the twist angle of the methyl C3v symmetry axis of trans-β-IEPOX relative to the surface is sensitive to the choice of DFT functional, the calculated tilt angle relative to the surface normal is largely independent of the functional and basis set. Moreover, we report that hybrid functionals such as B3LYP, ωB97X-D, PBE0, and B97-1 in combination with a modest basis set, such as 6-311G(d,p), provides good agreement with experimental data and much better performance than pure functionals such as PBE and BP86. However, improving the quality of the basis set only improves agreement with experimental data for calculations based on pure functionals. A conformational analysis, based on comparisons of calculated and experimental SFG spectra, suggests that trans-β-IEPOX points all of its oxygen atoms toward the silica/vapor interface.
Co-reporter:Sarah A. Saslow Gomez; Danielle Faurie-Wisniewski; Arlen Parsa; Jeff Spitz; Jennifer Amdur Spitz; Nancy C. Loeb
Journal of Chemical Education 2015 Volume 92(Issue 4) pp:638-642
Publication Date(Web):December 1, 2014
DOI:10.1021/ed500637y
The classroom exercise outlined here is a self-directed assignment that connects students to the environmental contamination problem surrounding the DePue Superfund site. By connecting chemistry knowledge gained in the classroom with a real-world problem, students are encouraged to personally connect with the problem while simultaneously developing skills in data management and interpretation. Designed for first-year undergraduate, general chemistry students, each of the four primary exercises builds upon the skills developed in those before it. This approach makes it easy to tailor this assignment to the needs of individual classrooms. This assignment was given as the first laboratory course assignment to a pilot group of 178 general chemistry undergraduate students at Northwestern University. Students enrolled in this course had already completed the first of two accelerated general chemistry courses. Over 75% of students scored in the 85% to 100% grade range, illustrating that the majority of students who completed this assignment demonstrated a high level of comprehension.
Co-reporter:Amanda L. Mifflin, Luis Velarde, Junming Ho, Brian T. Psciuk, Christian F. A. Negre, Carlena J. Ebben, Mary Alice Upshur, Zhou Lu, Benjamin L. Strick, Regan J. Thomson, Victor S. Batista, Hong-Fei Wang, and Franz M. Geiger
The Journal of Physical Chemistry A 2015 Volume 119(Issue 8) pp:1292-1302
Publication Date(Web):February 3, 2015
DOI:10.1021/jp510700z
Despite the importance of terpenes in biology, the environment, and catalysis, their vibrational spectra remain unassigned. Here, we present subwavenumber high-resolution broad-band sum frequency generation (HR-BB-SFG) spectra of the common terpene (+)-α-pinene that reveal 10 peaks in the C–H stretching region at room temperature. The high spectral resolution resulted in spectra with more and better resolved spectral features than those of the Fourier transform infrared, femtosecond stimulated Raman spectra in the bulk condensed phase and those of the conventional BB-SFG and scanning SFG spectroscopy of the same molecule on a surface. Experiment and simulation show the spectral line shapes with HR-BB-SFG to be accurate. Homogeneous vibrational decoherence lifetimes of up to 1.7 ps are assigned to specific oscillators and compare favorably to lifetimes computed from density functional tight binding molecular dynamics calculations. Phase-resolved spectra provided their orientational information. We propose the new spectroscopy as an attractive alternative to time domain vibrational spectroscopy or heterodyne detection schemes for studying vibrational energy relaxation and vibrational coherences in molecules at molecular surfaces or interfaces.
Co-reporter:Mona Shrestha, Yue Zhang, Mary Alice Upshur, Pengfei Liu, Sandra L. Blair, Hong-fei Wang, Sergey A. Nizkorodov, Regan J. Thomson, Scot T. Martin, and Franz M. Geiger
The Journal of Physical Chemistry A 2015 Volume 119(Issue 19) pp:4609-4617
Publication Date(Web):December 16, 2014
DOI:10.1021/jp510780e
The surfaces of secondary organic aerosol particles are notoriously difficult to access experimentally, even though they are the key location where exchange between the aerosol particle phase and its gas phase occurs. Here, we overcome this difficulty by applying standard and sub- 1 cm–1 resolution vibrational sum frequency generation (SFG) spectroscopy to detect C–H oscillators at the surfaces of secondary organic material (SOM) prepared from the ozonolysis of α-pinene at Harvard University and at the University of California, Irvine, that were subsequently collected on Teflon filters as well as CaF2 windows using electrostatic deposition. We find both samples yield comparable SFG spectra featuring an intense peak at 2940 cm–1 that are independent of spectral resolution and location or method of preparation. We hypothesize that the SFG spectra are due to surface-active C–H oscillators associated with the four-membered ring motif of α-pinene, which produces an unresolvable spectral continuum of approximately 50 cm–1 width reminiscent of the similar, albeit much broader, O–H stretching continuum observed in the SFG spectra of aqueous surfaces. Upon subjecting the SOM samples to cycles in relative humidity (RH) between <2% RH and ∼95% RH, we observe reversible changes in the SFG signal intensity across the entire spectral range surveyed for a polarization combination probing components of the vibrational transition dipole moments that are oriented parallel to the plane of incidence, but no signal intensity changes for any other polarization combination investigated. These results support the notion that the C–H oscillators at the surfaces of α-pinene-derived SOM deposited on CaF2 windows shift back and forth between two different molecular orientation distributions as the RH is lowered (more ordered) or raised (less ordered). The findings thus point toward the presence of a reversible surface switch for hindering (more ordered, <2%RH) and promoting (less ordered, ∼95%RH) exchange between the aerosol particle phase and its gas phase.
Co-reporter:Julianne M. Troiano
The Journal of Physical Chemistry C 2015 Volume 119(Issue 1) pp:534-546
Publication Date(Web):December 10, 2014
DOI:10.1021/jp512107z
This work presents molecular-level investigations of how well-characterized silica-supported phospholipid bilayers formed from either pure DOPC or a 9:1 mixture of DOPC:DOTAP interact with positively and negatively charged 4 nm gold metal nanoparticles at pH 7.4 and NaCl concentrations ranging from 0.001 to 0.1 M. Second harmonic generation (SHG) charge screening measurements indicate the supported bilayers carry a negative interfacial potential. Resonantly enhanced SHG measurements probing electronic transitions within the gold core of the nanoparticles show the particles interact irreversibly with the supported bilayers at a range of concentrations. At 0.1 M NaCl, surface coverages for the particles functionalized with the negatively charged ligand mercaptopropionic acid (MPA) or wrapped in the cationic polyelectrolyte poly(allylamine) hydrochloride (PAH) are estimated from a joint analysis of QCM-D, XPS, AFM, and ToF-SIMS to be roughly 1 × 107 and 1 × 1011 particles cm–2, respectively. Results from complementary SHG charge screening experiments point to the possibility that the surface coverage of the MPA-coated particles is more limited by interparticle Coulomb repulsion due to the charges within their hydrodynamic volumes than with the PAH-wrapped particles. Yet, SHG adsorption isotherms indicate that the interaction strength per particle is independent of ionic strength and particle coating, highlighting the importance of multivalent interactions. 1H NMR spectra of the lipids within vesicles suspended in solution show little change upon interaction with either particle type but indicate loosening of the gold-bound PAH polymer wrapping upon attachment to the vesicles. The thermodynamic, spectroscopic, and electrostatic data presented here may serve to benchmark experimental and computational studies of nanoparticle attachment processes at the nano–bio interface.
Co-reporter:Hilary M. Chase, Brian T. Psciuk, Benjamin L. Strick, Regan J. Thomson, Victor S. Batista, and Franz M. Geiger
The Journal of Physical Chemistry A 2015 Volume 119(Issue 14) pp:3407-3414
Publication Date(Web):March 16, 2015
DOI:10.1021/acs.jpca.5b02208
We combine deuterium labeling, density functional theory calculations, and experimental vibrational sum frequency generation spectroscopy into a form of “counterfactual-enabled molecular spectroscopy” for producing reliable vibrational mode assignments in situations where local group mode approximations are insufficient for spectral interpretation and vibrational mode assignments. We demonstrate the method using trans-β-isoprene epoxydiol (trans-β-IEPOX), a first-generation product of isoprene relevant to atmospheric aerosol formation, and one of its deuterium-labeled isotopologues at the vapor/silica interface. We use our method to determine that the SFG responses that we obtain from trans-β-IEPOX are almost exclusively due to nonlocal modes involving multiple C–H groups oscillating at the same frequency as one vibrational mode. We verify our assignments using deuterium labeling and use DFT calculations to predict SFG spectra of additional isotopologues that have not yet been synthesized. Finally, we use our new insight to provide a viable alternative to molecular orientation analysis methods that rely on local mode approximations in cases where the local mode approximation is not applicable.
Co-reporter:Merve Doğangün, Mimi N. Hang, Julianne M. Troiano, Alicia C. McGeachy, Eric S. Melby, Joel A. Pedersen, Robert J. Hamers, and Franz M. Geiger
ACS Nano 2015 Volume 9(Issue 9) pp:8755
Publication Date(Web):August 6, 2015
DOI:10.1021/acsnano.5b01440
Given the projected massive presence of redox-active nanomaterials in the next generation of consumer electronics and electric vehicle batteries, they are likely to eventually come in contact with cell membranes, with biological consequences that are currently not known. Here, we present nonlinear optical studies showing that lithium nickel manganese cobalt oxide nanosheets carrying a negative ζ-potential have no discernible consequences for lipid alignment and interleaflet composition in supported lipid bilayers formed from zwitterionic and negatively charged lipids. In contrast, lithiated and delithiated LiCoO2 nanosheets having positive and neutral ζ-potentials, respectively, alter the compositional asymmetry of the two membrane leaflets, and bilayer asymmetry remains disturbed even after rinsing. The insight that some cobalt oxide nanoformulations induce alterations to the compositional asymmetry in idealized model membranes may represent an important step toward assessing the biological consequences of their predicted widespread use.Keywords: nano−bio interface; phospholipid bilayer asymmetry; secondary ion mass spectrometry; silica/water interfaces; sum-frequency generation; transbilayer movement;
Co-reporter:Danielle Faurie-Wisniewski
The Journal of Physical Chemistry C 2014 Volume 118(Issue 40) pp:23256-23263
Publication Date(Web):September 10, 2014
DOI:10.1021/jp5069472
The synthesis and characterization of 15, 25, 50, and 70 nm thin iron films having chemical impurities below the detection limit of various analytical techniques is reported. As established herein, the films are chemically pure and formed by electron beam deposition from inexpensive and readily available iron sources of 3N5 purity. Chemical purity of the thin films was achieved using mean deposition rates of 0.3 nm/s or higher, at which point the melting point of iron is reached at the iron source surface and a shutter is opened, from which point on the rate of transfer of impurities present in the source to the target is low enough that they are not observed in the film as confirmed via X-ray photoelectron spectroscopy (XPS), reported here for energies between 0 and 1200 eV. Nanoindentation measurements indicate the iron films to be 14 times harder than bulk iron. The iron films are shown by XPS to be coated with a 3 nm thin overlayer of Fe3+, which is possibly present in the form of Fe3O4, even though other forms of iron oxide are likely to be present as well, as indicated by Raman and XPS spectroscopy. Grazing incidence angle X-ray diffraction experiments indicate the presence of crystalline Fe0 with low index faces exposed but no crystallinity of the iron oxide overlayer. Atomic force microscopy of the iron film surfaces indicates narrowing and shifts to lower heights in the height distribution of nanoscale features formed during the film deposition process as the film thickness decreases. Second harmonic generation is then used to determine that the interfacial charge density of the thinnest iron film is −0.007(3) C/m2 at pH 7.
Co-reporter:Jennifer L. Achtyl ; Ivan V. Vlassiouk ; Sheng Dai ;Franz Geiger
The Journal of Physical Chemistry C 2014 Volume 118(Issue 31) pp:17745-17755
Publication Date(Web):July 8, 2014
DOI:10.1021/jp5047547
The adsorption of 1-hexanol from cyclohexane-d12 at single-layer graphene/α-Al2O3 interfaces was probed at mole percent values as low as 0.05 in the C–H stretching region using vibrational sum frequency generation (SFG). The SFG spectra are indiscernible from those obtained in the absence of graphene, and from those obtained in the presence of graphene oxide films prepared via oxygen plasma treatment of pristine single-layer graphene. A Langmuir adsorption model yields observed free adsorption energies of −19.9(5) to −20.9(3) kJ/mol for the three interfaces. The results indicate that the molecular structure of the hexanol alkyl chain is subject to the same orientation distribution when graphene, oxidized or not, is present or absent at the α-Al2O3/cyclohexane-d12 interface. Moreover, it appears that the adsorption of 1-hexanol in this binary mixture is driven by hexanol interactions with the underlying oxide support, and that a single layer of graphene does not influence the extent of this interaction, even when defects are introduced to it. Finally, our structural and quantitative thermodynamic data provide important benchmarks for theoretical calculations and atomistic simulations of liquid/graphene interfaces. We hypothesize that defects emerging in graphene during operation of any device application that relies on layered solvent/graphene/oxide interfaces have little impact on the interfacial structure or thermodynamics, at least for the binary mixture and over the range of defect densities probed in our studies.
Co-reporter:David S. Jordan
The Journal of Physical Chemistry C 2014 Volume 118(Issue 50) pp:28970-28977
Publication Date(Web):January 8, 2014
DOI:10.1021/jp409852v
Second harmonic generation (SHG) and the χ(3) technique are used to investigate the effect of oxalic acid on the adsorption of aluminum [Al(III)] to the fused silica/water interface. Al(III) adsorption isotherms are measured in the presence and absence of oxalic acid and fit with the Diffuse Layer model to quantify thermodynamic binding parameters. The evolution of charge density throughout the adsorption process is analyzed and used to derive binding mechanisms at the molecular level. It is found that 0.5 mM oxalic acid irreversibly binds to and increases the negative surface charge on the fused silica surface −0.003 C/m2, corresponding to 1.9 × 1012 bound oxalate ions per cm2 under those conditions if oxalate were singly charged at the interface. Oxalic acid decreases the strength of the Al(III) adsorption interaction: the binding constant, Kads, decreases from 60 000 ± 10 000 to 1100 (200) M–1, corresponding to a reduction in the apparent adsorption free energy, ΔGads, from −37.2(4) to −27.3(5) kJ/mol, while surface charge densities at maximum metal coverage, σm, increase from 0.004(2) to 0.014(3) C/m2. Evidence is presented for a change in the dominant charge determining adsorption mechanism that corresponds with a change in Al(III) speciation. These results reveal important information about the surface activity and speciation of Al(III) in the presence of oxalic acid that have direct implications for the mobility, toxicity, and ultimate environmental fate of this metal pollutant.
Co-reporter:Sarah A. Saslow Gomez and Franz M. Geiger
The Journal of Physical Chemistry A 2014 Volume 118(Issue 46) pp:10974-10981
Publication Date(Web):October 21, 2014
DOI:10.1021/jp506283y
The interaction of Al(III), Sc(III), and La(III) with muscovite–water interfaces was studied at pH 4 and 10 mM NaCl using second harmonic generation (SHG) and X-ray photoelectron spectroscopy (XPS). SHG data for Sc(III) and La(III) suggest complete and/or partial irreversible adsorption that is attributed by XPS to the growth of Sc(III) and La(III) hydroxides/oxides on the muscovite surface. Al(III) adsorption appears to coincide with the growth of gibbsite (Al(OH)3) deposits on the muscovite surface, as indicated by the magnitude of the interfacial potential computed from the SHG data. This interpretation of the data is consistent with previous studies reporting the epitaxial growth of gibbsite on the muscovite surface under similar conditions. The implication of our findings is that the surface charge density of mica may change (and in the case of Al(III), even flip sign from negative (mica) to positive (gibbsite)) when Al(III), Sc(III), or La(III) is present in aqueous phases in contact with heterogeneous geochemical media rich in mica-class minerals, even at subsaturation conditions.
Co-reporter:Jennifer L. Achtyl, Ivan V. Vlassiouk, Sumedh P. Surwade, Pasquale F. Fulvio, Sheng Dai, and Franz M. Geiger
The Journal of Physical Chemistry B 2014 Volume 118(Issue 28) pp:7739-7749
Publication Date(Web):February 11, 2014
DOI:10.1021/jp410298e
This work reports thermodynamic and electrostatic parameters for fused silica/water interfaces containing cm2-sized graphene ranging from a single layer of pristine graphene to defected graphene. Second harmonic generation (SHG) measurements carried out at pH 7 indicate that the surface charge density of the fused silica/water interface containing the defected graphene (−0.009(3) to −0.010(3) C/m2) is between that of defect-free single layer graphene (−0.0049(8) C/m2) and bare fused silica (−0.013(6) C/m2). The interfacial free energy of the fused silica/water interface calculated from the Lippmann equation is reduced by a factor of 7 in the presence of single-layer pristine graphene, while defected graphene reduces it only by a factor of at most 2. Subsequent SHG adsorption isotherm studies probing the Mg2+ adsorption at the fused silica/water interface result in fully reversible metal ion interactions and observed binding constants, Kads, of 4(1) – 5(1) × 103 M–1 for pristine graphene and 3(1) – 4(1) × 103 M–1 for defected graphene, corresponding to adsorption free energies, ΔGads, referenced to the 55.5 molarity of water, of −30(1) to −31.1(7) kJ/mol for both interfaces, comparable to Mg2+ adsorption at the bare fused silica/water interface. Maximum Mg2+ ion densities are obtained from Gouy–Chapman model fits to the Langmuir adsorption isotherms and found to range from 1.1(5) – 1.5(4) × 1012 ions adsorbed per cm2 for pristine graphene and 2(1) – 3.1(5) × 1012 ions adsorbed per cm2 for defected graphene, slightly smaller than those of for Mg2+ adsorption at the bare fused silica/water interface ((2–4) × 1012 ions adsorbed per cm2), assuming the magnesium ions are bound as divalent species. We conclude that the presence of defects in the graphene sheet, which we estimate here to be around 1.3 × 1011 cm2, imparts only subtle changes in the thermodynamic and electrostatic parameters quantified here.
Co-reporter:Stephanie R. Walter ; Kaylie L. Young ; Joseph G. Holland ; Richard L. Gieseck ; Chad A. Mirkin
Journal of the American Chemical Society 2013 Volume 135(Issue 46) pp:17339-17348
Publication Date(Web):October 24, 2013
DOI:10.1021/ja406551k
Label-free studies carried out under aqueous phase conditions quantify the number of Mg2+ ions binding to surface-immobilized T40 sequences, the subsequent reordering of DNA on the surface, and the consequences of Mg2+ binding for DNA–DNA interactions. Second harmonic generation measurements indicate that, within error, 18–20 Mg2+ ions are bound to the T40 strand at saturation and that the metal–DNA interaction is associated with a near 30% length contraction of the strand. Structural reordering, evaluated using vibrational sum frequency generation, atomic force microscopy, and dynamic light scattering, is attributed to increased charge screening as the Mg2+ ions bind to the negatively charged DNA, reducing repulsive Coulomb forces between nucleotides and allowing the DNA single strands to collapse or coil upon themselves. The impact of Mg2+ binding on DNA hybridization and duplex stability is assessed with spherical nucleic acid (SNA) gold nanoparticle conjugates in order to determine an optimal working range of Mg2+ concentrations for DNA–DNA interactions in the absence of NaCl. The findings are consistent with a charge titration effect in which, in the absence of NaCl, (1) hybridization does not occur at room temperature if an average of 17.5 or less Mg2+ ions are bound per T40 strand, which is not reached until the bulk Mg2+ concentration approaches 0.5 mM; (2) hybridization proceeds, albeit with low duplex stability having an average Tm of 31(3)°C, if an average of 17.5–18.0 Mg2+ ions are bound; and (3) highly stable duplexes having a Tm of 64(2)°C form if 18.5–19.0 Mg2+ ions are bound, corresponding to saturation of the T40 strand.
Co-reporter:Jennifer L. Achtyl ; Ivan V. Vlassiouk ; Pasquale F. Fulvio ; Shannon M. Mahurin ; Sheng Dai
Journal of the American Chemical Society 2013 Volume 135(Issue 3) pp:979-981
Publication Date(Web):January 8, 2013
DOI:10.1021/ja3120899
Fluid/solid interfaces containing single-layer graphene are important in the areas of chemistry, physics, biology, and materials science, yet this environment is difficult to access with experimental methods, especially under flow conditions and in a label-free manner. Herein, we demonstrate the use of second harmonic generation to quantify the interfacial free energy at the fused silica/single-layer graphene/water interface at pH 7 and under conditions of flowing aqueous electrolyte solutions ranging in NaCl concentrations from 10–4 to 10–1 M. Our analysis reveals that single-layer graphene reduces the interfacial free energy density of the fused silica/water interface by a factor of up to 7, which is substantial given that many interfacial processes, including those that are electrochemical in nature, are exponentially sensitive to interfacial free energy density.
Co-reporter:Andrew P. Ault, Defeng Zhao, Carlena J. Ebben, Michael J. Tauber, Franz M. Geiger, Kimberly A. Prather and Vicki H. Grassian
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 17) pp:6206-6214
Publication Date(Web):19 Feb 2013
DOI:10.1039/C3CP43899F
Sea spray aerosol (SSA) represents one of the largest aerosol components in our atmosphere. SSA plays a major role in influencing climate; however the overall impacts remain poorly understood due to the overall chemical complexity. SSA is comprised of a mixture of inorganic and organic components in varying proportions that change as a function of particle size and seawater composition. In this study, nascent SSA particles were produced using breaking waves, resulting in compositions and sizes representative of the open ocean. The composition of individual SSA particles ranging in size from ca. 0.15 to 10 μm is measured using Raman microspectroscopy, while the interfacial composition of collections of size-resolved particles is probed by sum frequency generation (SFG). Raman spectra of single particles have bands in the 980 to 1030 cm−1 region associated with the symmetric stretch of the sulfate anion, the 2800 to 3000 cm−1 region associated with carbon–hydrogen stretches, and from 3200–3700 cm−1 associated with the oxygen–hydrogen stretches of water. The relative intensities of these features showed a strong dependence on particle size. In particular, submicrometer particles exhibited a larger amount of organic matter compared to supermicrometer particles. However, for external surfaces of homogeneous SSA particles (i.e. particles without a solid inclusion), and also the interfaces of mixed-phase particles, there was a strong SFG response in the aliphatic C–H stretching region for both sub- and supermicrometer particles. This finding suggests that organic material present in supermicrometer particles primarily resides at the interface. The presence of methylene contributions in the SFG spectra indicated disordered alkyl chains, in contrast to what one might expect for a surfactant layer on a sea salt particle. Changes in peak frequencies and relative intensities in the C–H stretching region are seen for some particles after the addition of bacteria, phytoplankton, and growth medium to the seawater. This study provides new insights into the bulk and surface composition of SSA particles and represents a step forward in our understanding of this globally abundant aerosol. It also provides insights into the development of model systems for SSA that may more accurately represent the organic layer at the surface.
Co-reporter:Carlena J. Ebben, Andrew P. Ault, Matthew J. Ruppel, Olivia S. Ryder, Timothy H. Bertram, Vicki H. Grassian, Kimberly A. Prather, and Franz M. Geiger
The Journal of Physical Chemistry A 2013 Volume 117(Issue 30) pp:6589-6601
Publication Date(Web):July 2, 2013
DOI:10.1021/jp401957k
We present vibrational sum frequency generation (SFG) spectra of the external surfaces and the internal interfaces of size-selected sea spray aerosol (SSA) particles generated at the wave flume of the Scripps Hydraulics Laboratory. Our findings support SSA particle models that invoke the presence of surfactants in the topmost particle layer and indicate that the alkyl chains of surfactant-rich SSA particles are likely to be disordered. Specifically, the SFG spectra suggest that across the range of sizes studied, surfactant-rich SSA particles contain CH oscillators that are subject to molecular orientation distributions that are broader than the narrow molecular distribution functions associated with well-ordered and well-aligned alkyl chains. This result is consistent with the interpretation that the permeability of organic layers at SSA particle surfaces to small reactive and nonreactive molecules may be substantial, allowing for much more exchange between reactive and nonreactive species in the gas or the condensed phase than previously thought. The SFG data also suggest that a one-component model is likely to be insufficient for describing the SFG responses of the SSA particles. Finally, the similarity of the SFG spectra obtained from the wave flume microlayer and 150 nm-sized SSA particles suggests that the SFG active CH oscillators in the topmost layer of the wave flume and the particle accumulation mode may be in similar chemical environments. Needs for additional research activities are discussed in the context of the results presented.
Co-reporter:Mona Shrestha, Yue Zhang, Carlena J. Ebben, Scot T. Martin, and Franz M. Geiger
The Journal of Physical Chemistry A 2013 Volume 117(Issue 35) pp:8427-8436
Publication Date(Web):July 22, 2013
DOI:10.1021/jp405065d
Secondary organic material (SOM) was produced in a flow tube from α-pinene ozonolysis, and collected particles were analyzed spectroscopically via a nonlinear coherent vibrational spectroscopic technique, namely sum frequency generation (SFG). The SOM precursor α-pinene was injected into the flow tube reactor at concentrations ranging from 0.125 ± 0.01 ppm to 100 ± 3 ppm. The oxidant ozone was varied from 0.15 ± 0.02 to 194 ± 2 ppm. The residence time was 38 ± 1 s. The integrated particle number concentrations, studied using a scanning mobility particle sizer (SMPS), varied from no particles produced up to (1.26 ± 0.02) × 107 cm–3 for the matrix of reaction conditions. The mode diameters of the aerosols increased from 7.7 nm (geometric standard deviation (gsd), 1.0) all the way to 333.8 nm (gsd, 1.9). The corresponding volume concentrations were as high as (3.0 ± 0.1) × 1014 nm3 cm–3. The size distributions indicated access to different particle growth stages, namely condensation, coagulation, or combination of both, depending on reaction conditions. For filter collection and subsequent spectral analysis, reaction conditions were selected that gave a mode diameter of 63 ± 3 nm and 93 ± 3 nm, respectively, and an associated mass concentration of 12 ± 2 μg m–3 and (1.2 ± 0.1) × 103 μg m–3 for an assumed density of 1200 kg m–3. Teflon filters loaded with 24 ng to 20 μg of SOM were analyzed by SFG. The SFG spectra obtained from particles formed under condensational and coagulative growth conditions were found to be quite similar, indicating that the distribution of SFG-active C–H oscillators is similar for particles prepared under both conditions. The spectral features of these flow-tube particles agreed with those prepared in an earlier study that employed the Harvard Environmental Chamber. The SFG intensity was found to increase linearly with the number of particles, consistent with what is expected from SFG signal production from particles, while it decreased at higher mass loadings of 10 and 20 μg, consistent with the notion that SFG probes the top surface of the SOM material following the complete coverage of the filter. The linear increase in SFG intensity with particle density also supports the notion that the average number of SFG active oscillators per particle is constant for a given particle size, that the particles are present on the collection filters in a random array, and that the particles are not coalesced. The limit of detection of SFG intensity was established as 24 ng of mass on the filter, corresponding to a calculated density of about 100 particles in the laser spot. As established herein, the technique is applicable for detecting low particle number or mass concentrations in ambient air. The related implication is that SFG is useful for short collection times and would therefore provide increased temporal resolution in a locally evolving atmospheric environment.
Co-reporter:Joseph G. Holland and Franz M. Geiger
The Journal of Physical Chemistry B 2013 Volume 117(Issue 3) pp:825-832
Publication Date(Web):December 11, 2012
DOI:10.1021/jp3105858
The binding of Y(III) ions to surface-immobilized single-stranded 20-mers of guanine was studied using the Eisenthal χ(3) technique and AFM. The free energy of binding for Y(III) to the G20 sequence was found to be −39.5(8) kJ/mol. Furthermore, yttrium binds much more strongly to surface-immobilized oligonucleotides than the divalent metals previously reported. At maximum surface coverage, Y(III) ion densities range between one to three ions bound per strand. Comparatively, Mg(II) binds to the G20-functionalized interface in much higher ion densities. This result may be explained, in part, by the larger hydration sphere radius of Y(III) compared to that of Mg(II). The ion loading and binding free energy results, in conjunction with other surface and bulk aqueous phase studies, suggest that a fully hydrated +2 or +3 yttrium ion binds to the oligonucleotides through an outer-sphere mechanism. Tapping mode AFM results indicate that oligonucleotide height does not appreciably decrease following Y(III) binding. These results, together with the low ion densities for Y(III) ions, indicate that Y(III) strand loading may not significantly decrease the intrastrand Coulombic repulsions in order to cause a significant decrease in oligomer height.
Co-reporter:David S. Jordan ; Christopher J. Hull ; Julianne M. Troiano ; Shannon C. Riha ; Alex B. F. Martinson ; Kevin M. Rosso
The Journal of Physical Chemistry C 2013 Volume 117(Issue 8) pp:4040-4047
Publication Date(Web):January 31, 2013
DOI:10.1021/jp3113057
Iron oxides are a ubiquitous class of compounds that are involved in many biological, geological, and technological processes, and the Fe(III)/Fe(II) redox couple is a fundamental transformation pathway; however, the study of iron oxide surfaces in aqueous solution by powerful spectroscopic techniques has been limited due to “strong absorber problem”. In this work, atomic layer deposition (ALD) thin films of polycrystalline α-Fe2O3 were analyzed using the Eisenthal χ(3) technique, a variant of second harmonic generation that reports on interfacial potentials. By determining the surface charge densities at multiple pH values, the point of zero charge was found to be 5.5 ± 0.3. The interaction of aqueous Fe(II) at pH 4 and in 1 mM NaCl with ALD-prepared hematite was found to be fully reversible and to lead to about 4 times more ferrous iron ions adsorbed per square centimeter than on fused-silica surfaces under the same conditions. The data are consistent with a recently proposed conceptual model for net Fe(II) uptake or release that is underlain by a dynamic equilibrium between Fe(II) adsorbed onto hematite, electron transfer into favorable surface sites with attendant Fe(III) deposition, and electron conduction to favorable remote sites that release and replenish aqueous Fe(II).
Co-reporter:Julianne M. Troiano ; David S. Jordan ; Christopher J. Hull
The Journal of Physical Chemistry C 2013 Volume 117(Issue 10) pp:5164-5171
Publication Date(Web):February 18, 2013
DOI:10.1021/jp3122819
The fate of chromium in the environment relies heavily on its redox chemistry and interaction with iron oxide surfaces. Atomic layer deposition was used to deposit a 10 nm film of polycrystalline α-Fe2O3 (hematite) onto a fused silica substrate which was analyzed using second harmonic generation (SHG), a coherent, surface-specific, nonlinear optical technique. Specifically, the χ(3) technique was used to investigate the adsorption of Cr(III) and Cr(VI) to the hematite/water interface under flow conditions at pH 4 with 10 mM NaCl. We observed partially irreversible adsorption of Cr(III), the extent of which was found to be dependent on the concentration of Cr(III) ions in solution. This result was confirmed using X-ray photoelectron spectroscopy. The interaction of Cr(III) with hematite is compared with the adsorption of Cr(III) to the silica/water interface, which is the substrate for the ALD-prepared hematite films, and found to be fully reversible under the same experimental conditions. The observed binding constant for Cr(III) interacting with the silica surface was found to be 4.0(6) × 103 M–1, which corresponds to an adsorption free energy of −30.5(4) kJ/mol when referenced to 55.5 M water. The surface charge density at maximum metal ion surface coverage was found to be 0.005(1) C/m2, which corresponds to 1.0 × 1012 ions/cm2 assuming a +3 charge for chromium. In contrast, the observed binding constant for Cr(III) interacting reversibly with the hematite surface was calculated to be 2(2) × 104 M–1, corresponding to an adsorption free energy of −35(2) kJ/mol when referenced to 55.5 M water. The surface charge density at maximum metal ion surface coverage was found to be 0.004(5) C/m2 for the reversibly bound chromium species, which corresponds to 8.3 × 1011 reversibly bound ions per cm2, again assuming a +3 charge of chromium. The data also allows us to estimate that about 6.7 × 1012 Cr(III) ions are irreversibly bound per cm2 hematite at saturation coverage. The results of this investigation suggest that the use of hematite in permeable reactive barriers, for cost-effective chromium remediation, allows for Cr(III) remediation at very low concentrations through adsorptive and redox processes but quickly renders the barriers ineffective at high chromium concentrations due to surface saturation.
Co-reporter:Stephanie R. Walter ; Jangdae Youn ; Jonathan D. Emery ; Sumit Kewalramani ; Jonathan W. Hennek ; Michael J. Bedzyk ; Antonio Facchetti ; Tobin J. Marks
Journal of the American Chemical Society 2012 Volume 134(Issue 28) pp:11726-11733
Publication Date(Web):June 18, 2012
DOI:10.1021/ja3036493
Organic thin film transistor (OTFT) performance is highly materials interface-dependent, and dramatic performance enhancements can be achieved by properly modifying the semiconductor/gate dielectric interface. However, the origin of these effects is not well understood, as this is a classic “buried interface” problem that has traditionally been difficult to address. Here we address the question of how n-octadecylsilane (OTS)–derived self-assembled monolayers (SAMs) on Si/SiO2 gate dielectrics affect the OTFT performance of the archetypical small-molecule p-type semiconductors P-BTDT (phenylbenzo[d,d]thieno[3,2-b;4,5-b]dithiophene) and pentacene using combined in situ sum frequency generation spectroscopy, atomic force microscopy, and grazing incidence and reflectance X-ray scattering. The molecular order and orientation of the OTFT components at the dielectric/semiconductor interface is probed as a function of SAM growth mode in order to understand how this impacts the overlying semiconductor growth mode, packing, crystallinity, and carrier mobility, and hence, transistor performance. This understanding, using a new, humidity-specific growth procedure, leads to a reproducible, scalable process for highly ordered OTS SAMs, which in turn nucleates highly ordered p-type semiconductor film growth, and optimizes OTFT performance. Surprisingly, the combined data reveal that while SAM molecular order dramatically impacts semiconductor crystalline domain size and carrier mobility, it does not significantly influence the local orientation of the overlying organic semiconductor molecules.
Co-reporter:Sarah A. Saslow Gomez, David S. Jordan, Julianne M. Troiano, and Franz M. Geiger
Environmental Science & Technology 2012 Volume 46(Issue 20) pp:11154-11161
Publication Date(Web):September 11, 2012
DOI:10.1021/es302879y
Uranyl adsorption at the muscovite (mica)/water interface was studied by second harmonic generation (SHG). Using the nonresonant χ3 technique and the Gouy–Chapman model, the initial surface charge density of the mica surface was determined to be −0.022(1) C/m2 at pH 6 and in the presence of dissolved carbonate. Under these same conditions, uranyl adsorption isotherms collected using nonresonant χ3 experiments and resonantly enhanced SHG experiments that probe the ligand-to-metal charge transfer bands of the uranyl cation yielded a uranyl binding constant of 3(1) × 107 M–1, corresponding to a Gibbs free energy of adsorption of −52.6(8) kJ/mol, and a maximum surface charge density at monolayer uranyl coverage of 0.028(3) C/m2. These results suggest favorable adsorption of uranyl ions to the mica interface through strong ion-dipole or hydrogen interactions, with a 1:1 uranyl ion to surface site ratio that is indicative of monovalent cations ((UO2)3(OH)5+, (UO2)4(OH)7+, UO2OH+, UO2Cl+, UO2(CH3COO–)+) binding at the interface, in addition to neutral uranyl species (UO2(OH)2 and UO2CO3). This work provides benchmark measurements to be used in the improvement of contaminant transport modeling.
Co-reporter:Ehow H. Chen, Sarah A. Saslow, SonBinh T. Nguyen, and Franz M. Geiger
The Journal of Physical Chemistry C 2012 Volume 116(Issue 12) pp:7016-7020
Publication Date(Web):March 5, 2012
DOI:10.1021/jp212331x
The interaction of Zn2+ ions with undecanol-functionalized fused silica/water interfaces was studied directly at the aqueous/solid interface. We characterized the surface functionalization using vibrational sum frequency generation (SFG) and X-ray photoelectron spectroscopy (XPS). We then employed the SHG χ(3) technique to determine the degree of silane functionalization, track Zn2+ adsorption directly at the hydroxyl-terminated undecanol silane-functionalized fused silica/aqueous interface at pH 7 and 10 mM NaCl concentration, determine the electrostatic and thermodynamic binding parameters, quantify the change in interfacial potential upon zinc ion adsorption, and compare these values to our previous work with glucosamine-functionalized and bare fused silica/water interfaces. The results from the calculated adsorption free energy suggest that 2:1 hydroxyl/metal coordination complexes, which have not been observed with natural carbohydrates in the bulk aqueous phase, are possible in interfacial environments, with direct implications for controlling and predicting coordination chemistry.
Co-reporter:Joseph G. Holland and Franz M. Geiger
The Journal of Physical Chemistry B 2012 Volume 116(Issue 22) pp:6302-6310
Publication Date(Web):May 9, 2012
DOI:10.1021/jp301573g
The binding of magnesium ions to surface-bound single-stranded oligonucleotides was studied under aqueous conditions using second harmonic generation (SHG) and atomic force microscopy (AFM). The effect of strand length on the number of Mg(II) ions bound and their free binding energy was examined for 5-, 10-, 15-, and 20-mers of adenine and guanine at pH 7, 298 K, and 10 mM NaCl. The binding free energies for adenine and guanine sequences were calculated to be −32.1(4) and −35.6(2) kJ/mol, respectively, and invariant with strand length. Furthermore, the ion density for adenine oligonucleotides did not change as strand length increased, with an average value of 2(1) ions/strand. In sharp contrast, guanine oligonucleotides displayed a linear relationship between strand length and ion density, suggesting that cooperativity is important. This data gives predictive capabilities for mixed strands of various lengths, which we exploit for 20-mers of adenines and guanines. In addition, the role sequence order plays in strands of hetero-oligonucleotides was examined for 5′-A10G10-3′, 5′-(AG)10-3′, and 5′-G10A10-3′ (here the -3′ end is chemically modified to bind to the surface). Although the free energy of binding is the same for these three strands (averaged to be −33.3(4) kJ/mol), the total ion density increases when several guanine residues are close to the 3′ end (and thus close to the solid support substrate). To further understand these results, we analyzed the height profiles of the functionalized surfaces with tapping-mode atomic force microscopy (AFM). When comparing the average surface height profiles of the oligonucleotide surfaces pre- and post- Mg(II) binding, a positive correlation was found between ion density and the subsequent height decrease following Mg(II) binding, which we attribute to reductions in Coulomb repulsion and strand collapse once a critical number of Mg(II) ions are bound to the strand.
Co-reporter:Jennifer L. Achtyl, Avram M. Buchbinder, and Franz M. Geiger
The Journal of Physical Chemistry Letters 2012 Volume 3(Issue 3) pp:280-282
Publication Date(Web):January 6, 2012
DOI:10.1021/jz2016796
The ability to study the interactions of hydrocarbons on carbon surfaces is an integral step toward gaining a molecular level understanding of the chemical reactions and physical properties occurring on them. Here, we apply vibrational sum frequency generation (SFG) to determine the tilt angle of toluene, a common organic solvent, on millimeter-thick highly oriented pyrolytic graphite (HOPG). The combination of a time-delay technique, which results in the successful suppression of the nonresonant SFG response, and a null angle method is shown to overcome the “strong optical absorber” problem posed by macroscopically thick carbon samples and yields a molecular tilt angle of toluene in the range of 37° to 42° from the surface normal. The implications of this approach for determining the orientation of organic species adsorbed on carbon interfaces, which are important for energy-relevant processes, are discussed.Keywords: carbon; graphite; HOPG; nonlinear optics; orientation; sum frequency generation; toluene;
Co-reporter:Ehow H. Chen, Stephanie R. Walter, SonBinh T. Nguyen, and Franz M. Geiger
The Journal of Physical Chemistry C 2012 Volume 116(Issue 37) pp:19886-19892
Publication Date(Web):August 23, 2012
DOI:10.1021/jp306437b
The conversion of surface-bound aminophenyl groups to azidophenyl moieties on SiOx surfaces was investigated as part of a mild, simple two-step strategy for “click”-based” surface functionalization with acetylene-functionalized reagents. Small terminal alkynes (phenylacetylene, 1-hexyne) and acetylene-modified single-stranded DNA 20-mers (T20) were then used as model compounds to test the efficiency of the 1,3-dipolar cycloaddition reaction. The identities of surface species were verified, and their coverages were quantified using X-ray photoelectron spectroscopy in the C 1s, N 1s, F 1s, Cl 2p, and P 2p regions. Depending on conditions, the yield of the azidification was in the 30–90% range, and the efficiency of triazole formation depended significantly on the rigidity of the acetylene reactant. Vibrational sum frequency generation was applied to probe the C–H stretching region and test the platform’s viability for minimizing spectral interference in the C–H stretching region. Fluorescence spectroscopy was also performed to verify the presence of fluorescein-tagged DNA single strands that have been coupled to the surface, while label-free DNA hybridization studies by vibrational sum frequency generation spectroscopy readily show the occurrence of duplex formation. Our results suggest that the two-step azidification–click sequence is a viable strategy for readily functionalizing silica and glass surfaces with molecules spanning a wide range of chemical complexity, including biopolymers.
Co-reporter:Avram M. Buchbinder ; Natalie A. Ray ; Junling Lu ; Richard P. Van Duyne ; Peter C. Stair ; Eric Weitz
Journal of the American Chemical Society 2011 Volume 133(Issue 44) pp:17816-17823
Publication Date(Web):September 15, 2011
DOI:10.1021/ja2067274
This work characterizes the adsorption, structure, and binding mechanism of oxygenated organic species from cyclohexane solution at the liquid/solid interface of optically flat alumina-supported palladium nanoparticle surfaces prepared by atomic layer deposition (ALD). The surface-specific nonlinear optical vibrational spectroscopy, sum-frequency generation (SFG), was used as a probe for adsorption and interfacial molecular structure. 1-Hexanoic acid is an overoxidation product and possible catalyst poison for the aerobic heterogeneous oxidation of 1-hexanol at the liquid/solid interface of Pd/Al2O3 catalysts. Single component and competitive adsorption experiments show that 1-hexanoic acid adsorbs to both ALD-prepared alumina surfaces and alumina surfaces with palladium nanoparticles, that were also prepared by ALD, more strongly than does 1-hexanol. Furthermore, 1-hexanoic acid adsorbs with conformational order on ALD-prepared alumina surfaces, but on surfaces with palladium particles the adsorbates exhibit relative disorder at low surface coverage and become more ordered, on average, at higher surface coverage. Although significant differences in binding constant were not observed between surfaces with and without palladium nanoparticles, the palladium particles play an apparent role in controlling adsorbate structures. The disordered adsorption of 1-hexanoic acid most likely occurs on the alumina support, and probably results from modification of binding sites on the alumina, adjacent to the particles. In addition to providing insight on the possibility of catalyst poisoning by the overoxidation product and characterizing changes in its structure that result in only small adsorption energy changes, this work represents a step toward using surface science techniques that bridge the complexity gap between fundamental studies and realistic catalyst models.
Co-reporter:Joseph G. Holland ; Jessica N. Malin ; David S. Jordan
Journal of the American Chemical Society 2011 Volume 133(Issue 8) pp:2567-2570
Publication Date(Web):February 3, 2011
DOI:10.1021/ja107883x
This article reports nonlinear optical measurements that quantify, for the first time directly and without labels, how many Mg2+ cations are bound to DNA 21-mers covalently linked to fused silica/water interfaces maintained at pH 7 and 10 mM NaCl, and what the thermodynamics are of these interactions. The overall interaction of Mg2+ with adenine, thymine, guanine, and cytosine is found to involve −10.0 ± 0.3, −11.2 ± 0.3, −14.0 ± 0.4, and −14.9 ± 0.4 kJ/mol, and nonspecific interactions with the phosphate and sugar backbone are found to contribute −21.0 ± 0.6 kJ/mol for each Mg2+ ion bound. The specific and nonspecific contributions to the interaction energy of Mg2+ with oligonucleotide single strands is found to be additive, which suggests that within the uncertainty of these surface-specific experiments, the Mg2+ ions are evenly distributed over the oligomers and not isolated to the most strongly binding nucleobase. The nucleobases adenine and thymine are found to bind only three Mg2+ ions per 21-mer oligonucleotide, while the bases cytosine and guanine are found to bind eleven Mg2+ ions per 21-mer oligonucleotide.
Co-reporter:Matthew T. Frederick ; Jennifer L. Achtyl ; Kathryn E. Knowles ; Emily A. Weiss
Journal of the American Chemical Society 2011 Volume 133(Issue 19) pp:7476-7481
Publication Date(Web):April 22, 2011
DOI:10.1021/ja200466z
This Article reports measurements of the intra- and intermolecular ordering of tight-binding octylphosphonate ligands on the surface of colloidal CdSe quantum dots (QDs) within solid state films, and the dependence of this order on the size of the QDs. The order of the organic ligands, as probed by vibrational sum frequency generation (SFG) spectroscopy, decreases as the radius of the QDs decreases; this decrease is correlated with a decrease in the order of underlying Cd2+, as detected by X-ray photoelectron spectroscopy (XPS) line width measurements, for radii of the QDs, R > 2.4 nm, and is independent of the disorder of the Cd2+ for R < 2.4 nm. We believe that, for R < 2.4, the decreasing order of the ligands with decreasing size is due to an increase in the curvature of the QD surfaces. Disorder in the Cd2+ results from the presence of a shell of Cd2+–surfactant complexes that form during synthesis, so this work demonstrates the possibility for chemical control over molecular order within films of colloidal QDs by changing the surfactant mixture.
Co-reporter:Joseph G. Holland, David S. Jordan, and Franz M. Geiger
The Journal of Physical Chemistry B 2011 Volume 115(Issue 25) pp:8338-8345
Publication Date(Web):May 25, 2011
DOI:10.1021/jp202884n
The binding of Sr(II), Ca(II), Mg(II), Ba(II), Mn(II), Zn(II), and Cd(II) to silica/water interfaces functionalized with A15T6 oligonucleotides was quantified at pH 7 and 10 mM NaCl using the Eisenthal χ(3) technique. The binding free energies range from −31.1(6) kJ/mol for Ba(II) to −33.8(4) kJ/mol for Ca(II). The ion densities were found to range from 2(1) ions/strand for Zn(II) to 11(1) ions/strand for Cd(II). Additionally, we quantified Mg(II) binding in the presence of varying background electrolyte concentrations which showed that the binding free energies changed in a linear fashion from −39.3(8) to −27(1) kJ/mol over the electrolyte concentration range of 1–80 mM, respectively. An adsorption free energy versus interfacial potential analysis allowed us to elucidate the speciation of the bound Mg(II) ions and to identify three possible binding pathways. Our findings suggest that Mg(II) binds as a fully hydrated divalent cation, most likely displacing DNA-bound Na ions. These measurements will serve as a benchmark for computer simulations of divalent metal cation/DNA interactions for geochemical and biosensing applications.
Co-reporter:Avram M. Buchbinder ; Julianne M. Gibbs-Davis ; Grace Y. Stokes ; Mark D. Peterson ; Eric Weitz
The Journal of Physical Chemistry C 2011 Volume 115(Issue 37) pp:18284-18294
Publication Date(Web):August 11, 2011
DOI:10.1021/jp205912h
A method is proposed and tested in which vibrational sum frequency generation (SFG) spectra are assigned to vibrational modes in cyclic hydrocarbon species at transparent oxide surfaces. This method is especially effective for identifying asymmetric C–H stretches which are particularly difficult to assign in cyclic hydrocarbons because they have frequencies similar to those of overtones and Fermi resonances, and because multiple nondegenerate asymmetric C–H stretches occur. The ratios between SFG signal intensities in two different experimental polarization combinations are compared to the predicted values assuming local C2v symmetry for an asymmetric CH2 stretching vibration. By applying this method to silica surfaces containing covalently linked cyclopropyl, cyclobutyl, cyclopentyl, and cyclopentenyl groups with fixed orientations, and benchmarking to published gas phase spectra, the observed SFG peaks are assigned to either (1) isolated asymmetric stretches of CH2 groups possessing local C2v symmetry, (2) motions of CH bonds coupled throughout the molecule not possessing local C2v symmetry, or (3) symmetric stretching or other C–H stretching motions. The SFG spectra of the less-strained five-membered rings contain peaks that—based on polarization ratios—correspond to isolated asymmetric stretches. For these species, the results contradict a full-molecule normal mode description of molecular vibrations.
Co-reporter:Patrick L. Hayes, Jessica N. Malin, David S. Jordan, Franz M. Geiger
Chemical Physics Letters 2010 Volume 499(4–6) pp:183-192
Publication Date(Web):29 October 2010
DOI:10.1016/j.cplett.2010.09.060
Abstract
This Letter provides a tutorial review of how the Eisenthal χ(3) Technique can be applied as a general method for quantifying (1) the amount of metal ions adsorbed, (2) the electrostatics associated with the adsorption processes, and (3) the thermodynamics, kinetics, and molecular mechanisms that underlie the interaction of the metal ions with aqueous/solid interfaces. We focus on our recent effort to quantify potentials at aqueous/solid interfaces in the presence of magnesium, calcium, strontium, and barium cations and determine alkaline earth metal ion adsorption thermodynamics at pH 7 and 10 mM NaCl. Important theoretical considerations for this method are discussed as well.
Co-reporter:Stephanie R. Walter and Franz M. Geiger
The Journal of Physical Chemistry Letters 2010 Volume 1(Issue 1) pp:9-15
Publication Date(Web):November 5, 2009
DOI:10.1021/jz9001086
The field of nonlinear optics continues to expand and surprise. The ability to study DNA with nonlinear optics has opened the door to understand, on a molecular level and without the use of external labels, the physical and chemical properties of DNA single and double strands at surfaces and interfaces. In this Perspective, we survey how nonlinear optical probes access the electronic, vibrational, electrostatic, and chiral signatures of interfacial DNA in its native state. We also show how this exciting new field is directly applicable to tracking and understanding molecular recognition in DNA oligonucleotides and aptamers.
Co-reporter:Ehow H. Chen ; Patrick L. Hayes ; SonBinh T. Nguyen
The Journal of Physical Chemistry C 2010 Volume 114(Issue 45) pp:19483-19488
Publication Date(Web):October 25, 2010
DOI:10.1021/jp108390p
The adsorption of Zn2+ to glucosamide-functionalized fused silica/water interfaces is studied using second harmonic generation (SHG). We characterize each step of the surface functionalization using vibrational sum frequency generation (SFG) and X-ray photoelectron spectroscopy (XPS), where specific vibrational modes in the C−H region and binding energies in the C1s and N1s region are determined to be indicative of glucose covalently tethered to the surface. We employ the SHG χ(3) technique to track Zn2+ adsorption and desorption directly at the glucosamide-functionalized fused silica/aqueous interface at pH 7 and 10 mM NaCl and determine the electrostatic and thermodynamic binding parameters using standard electrical double layer models to quantify the change in interfacial potential upon zinc adsorption. The results presented here allow for the possibility of 2:1 to 3:1 carbohydrate:metal coordination complexes and suggest the possibility for multivalent interactions which have not been observed with glucose in the bulk aqueous phase, where 1:1 complexes dominate. These findings suggest that interactions between metal ions and carbohydrate arrays may be much stronger at interfaces as opposed to the bulk phase, with direct implications for controlling and predicting coordination chemistry.
Co-reporter:Patrick L. Hayes, Alison R. Keeley and Franz M. Geiger
The Journal of Physical Chemistry B 2010 Volume 114(Issue 13) pp:4495-4502
Publication Date(Web):March 11, 2010
DOI:10.1021/jp911116q
Utilizing vibrational sum frequency generation (SFG), we characterized the structure of adsorbed cetyltrimethylammonium chloride (CTAC) at the silica/aqueous interface in the presence of 10 to 500 mM NaCl and as a function of surfactant surface coverage. For low ionic strengths (10 mM NaCl), results indicate that adsorbed aggregates do not change conformation with increasing surface coverage. Instead, the surfactant adsorbs as micelle-like structures at concentrations considerably lower than surface saturation and the CMC. At high ionic strengths (300−500 mM NaCl), the structure of the adlayer is considerably different: The SFG results indicate that for 30 μM bulk CTAC the surfactant packs with fewer gauche defects in the hydrocarbon backbone, which is attributed to reduced Coulomb repulsion between the positively charged surfactant headgroups, and the results also indicate that CTAC adsorbs as monomers at low surface coverage but then rearranges into aggregates at higher surface coverage.
Co-reporter:Grace Y. Stokes, Avram M. Buchbinder, Julianne M. Gibbs-Davis, Karl A. Scheidt, Franz M. Geiger
Vibrational Spectroscopy 2009 Volume 50(Issue 1) pp:86-98
Publication Date(Web):26 May 2009
DOI:10.1016/j.vibspec.2008.08.003
Using tailor-made organic compounds tethered to solid substrates through organo-silane chemistry, we present a reductionist model study aimed at understanding the mechanisms of heterogeneous organic oxidation reactions at solid/air interfaces. The surface vibrational spectra of glass slides functionalized with the tropospherically relevant olefins 1-pentene, 2-hexene, cyclopentene, cyclohexene, and a menthenol derivative via aniline-silane linkers have been obtained through polarization-resolved broadband vibrational sum frequency generation (SFG). The olefinic and aliphatic CH stretches located above and below 3000 cm−1, respectively, are clearly discernable and their spectral intensities are used to track, with 10 s time resolution, CC double bond oxidation reactions of surface-bound cyclohexene at room temperature and with low ppm amounts of ozone at 1 atm. The olefinic CH stretching mode disappears at a rate of 0.05(1) s−1, and the aliphatic asymmetric stretch modes increase at a rate of 0.04(1) s−1. Analogous experiments show the formation of methyl groups even for those olefins under investigation that do not originally possess methyl groups. The implications for heterogeneous organic oxidation chemistry involving tropospheric dust particles are discussed.
Co-reporter:Joshua A. Kellar, Jui-Ching Lin, Jun-Hyun Kim, Nathan L. Yoder, Kirk H. Bevan, Grace Y. Stokes, Franz M. Geiger, SonBinh T. Nguyen, Michael J. Bedzyk and Mark C. Hersam
The Journal of Physical Chemistry C 2009 Volume 113(Issue 7) pp:2919-2927
Publication Date(Web):2017-2-22
DOI:10.1021/jp8100249
Highly conjugated molecules bound to silicon are promising candidates for organosilicon electronic devices and sensors. In this study, 1-bromo-4-ethynylbenzene was synthesized and reacted with a hydrogen-passivated Si(111) surface via ultraviolet irradiation. Through an array of characterization and modeling tools, the binding configuration and morphology of the reacted molecule were thoroughly analyzed. Atomic force microscopy confirmed an atomically flat surface morphology following reaction, while X-ray photoelectron spectroscopy verified reaction to the surface via the terminal alkyne moiety. In addition, synchrotron X-ray characterization, including X-ray reflectivity, X-ray fluorescence, and X-ray standing wave measurements, enabled sub-angstrom determination of the position of the bromine atom with respect to the silicon lattice. This structural characterization was quantitatively compared with density functional theory (DFT) calculations, thus enabling the π-conjugation of the terminal carbon atoms to be deduced. The X-ray and DFT results were additionally corroborated with the vibrational spectrum of the organic adlayer, which was measured with sum frequency generation. Overall, these results illustrate that the terminal carbon atoms in 1-bromo-4-ethynylbenzene adlayers on Si(111) retain π-conjugation, thus revealing alkyne molecules as promising candidates for organosilicon electronics and sensing.
Co-reporter:Grace Y. Stokes, Ehow H. Chen, Stephanie R. Walter and Franz M. Geiger
The Journal of Physical Chemistry A 2009 Volume 113(Issue 31) pp:8985-8993
Publication Date(Web):July 10, 2009
DOI:10.1021/jp904104s
Important mechanistic differences regarding C═C double-bond oxidation processes under ozone-limited and ozone-rich reaction conditions for cyclohexene-functionalized fused silica substrates serving as model systems for studying heterogeneous C═C double bond oxidation chemistry in the troposphere are evaluated. By using broadband vibrational sum frequency generation (SFG), we track heterogeneous ozone reactions in real time. Ozone levels span three orders of magnitude and represent environments ranging from pristine remote continental regions to highly polluted urban centers, ranging from 30 ppb to 3 ppm (from 7 × 1011 molecules cm−3 to 7 × 1013 molecules cm−3). We determine reaction rates and reactive uptake coefficients (γ values). At these tropospherically relevant ozone levels, the heterogeneous reaction rates follow a Langmuir−Hinshelwood-type mechanism. The product formation rates, which we determine as a function of ozone concentrations, are found to be half of the olefin reaction rates. This ratio is consistent with the previously proposed reaction pathway involving the breaking of one C═C double bond containing two olefinic CH moieties to form a product containing only one methyl group and one polar carbonyl moiety. Contact angle histograms show that out of a total of 60 measurements, there are about 25 more measurements with contact angles up to ten degrees below the mean recorded prior to reaction when ozone levels resemble remote continental conditions (50 ppb) than when ozone levels resemble urban conditions (1 ppm). The implication of these results are that the methyl formation pathway in heterogeneous ozonolysis may be less favorable than the carboxylic acid- and secondary ozonide-production pathway for ozone-limited conditions (i.e., in the remote continental troposphere or during urban nighttime) as opposed to ozone-rich (i.e., polluted urban atmosphere) conditions.
Co-reporter:Patrick L. Hayes, Ehow H. Chen, Jennifer L. Achtyl and Franz M. Geiger
The Journal of Physical Chemistry A 2009 Volume 113(Issue 16) pp:4269-4280
Publication Date(Web):March 23, 2009
DOI:10.1021/jp810891v
Electrostatics and counterions play important roles in many supramolecular processes, including surfactant adsorption and aggregation at interfaces. Here, we assess their influence on how the common surfactant cetyltrimethylammonium (CTA) interacts with fused silica/aqueous interfaces by determining thermodynamic, kinetic, and electrostatic parameters for CTA adsorption across a range of NaCl concentrations (10−500 mM NaCl) using second harmonic generation (SHG). Using vibrational sum frequency generation (SFG), we demonstrate that vibrationally resonant contributions and nonresonant background contributions to the SFG signal intensity that depend on the interfacial potential can be quantified simultaneously during the adsorption process, which provides insight into the nonequilibrium dynamics of CTA adsorption. By analyzing the adsorption free energies as a function of interfacial potential at these four salt concentrations, the charge density per adsorbate is determined, indicating that CTA coadsorbs with counterions at a ratio of approximately 4 to 3 (i.e., 4 CTA+ ions for every 3 Cl− ion). The chemical (i.e., non-Coulombic) portion of the free energy is found to dominate the overall free energy of adsorption, indicating that CTA adsorption at these ionic strengths is primarily driven by the favorable hydrophobic interactions between interdigitated surfactant hydrocarbon chains in the adsorbed aggregate. By applying Gouy−Chapman−Stern theory to our data, an average charge density of 2.8(3) × 1013 charges/cm2, which corresponds to 0.7 to 1.7 molecules/nm2, was obtained for the four NaCl concentrations.
Co-reporter:Grace Y. Stokes, Avram M. Buchbinder, Julianne M. Gibbs-Davis, Karl A. Scheidt and Franz M. Geiger
The Journal of Physical Chemistry A 2008 Volume 112(Issue 46) pp:11688-11698
Publication Date(Web):October 22, 2008
DOI:10.1021/jp803277s
We report vibrational sum frequency generation (SFG) spectra of glass surfaces functionalized with 1-pentene, 2-hexene, cyclopentene, cyclohexene, and a menthenol derivative. The heterogeneous reactions of ozone with hydrocarbons covalently linked to oxide surfaces serve as models for studying heterogeneous oxidation of biogenic terpenes adsorbed to mineral aerosol surfaces commonly found in the troposphere. Vibrational SFG is also used to track the C═C double bond oxidation reactions initiated by ozone in real time and to characterize the surface-bound product species. Combined with contact angle measurements carried out before and after ozonolysis, the kinetic and spectroscopic studies presented here suggest reaction pathways involving vibrationally hot Criegee intermediates that compete with pathways that involve thermalized surface species. Kinetic measurements suggest that the rate limiting step in the heterogeneous C═C double bond oxidation reactions is likely to be the formation of the primary ozonide. From the determination of the reactive uptake coefficients, we find that ozone molecules undergo between 100 and 10000 unsuccessful collisions with C═C double bonds before the reaction occurs. The magnitude of the reactive uptake coefficients for the cyclic and linear olefins studied here does not follow the corresponding gas-phase reactivities but rather correlates with the accessibility of the C═C double bonds at the surface.
Co-reporter:Andrew P. Ault, Defeng Zhao, Carlena J. Ebben, Michael J. Tauber, Franz M. Geiger, Kimberly A. Prather and Vicki H. Grassian
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 17) pp:NaN6214-6214
Publication Date(Web):2013/02/19
DOI:10.1039/C3CP43899F
Sea spray aerosol (SSA) represents one of the largest aerosol components in our atmosphere. SSA plays a major role in influencing climate; however the overall impacts remain poorly understood due to the overall chemical complexity. SSA is comprised of a mixture of inorganic and organic components in varying proportions that change as a function of particle size and seawater composition. In this study, nascent SSA particles were produced using breaking waves, resulting in compositions and sizes representative of the open ocean. The composition of individual SSA particles ranging in size from ca. 0.15 to 10 μm is measured using Raman microspectroscopy, while the interfacial composition of collections of size-resolved particles is probed by sum frequency generation (SFG). Raman spectra of single particles have bands in the 980 to 1030 cm−1 region associated with the symmetric stretch of the sulfate anion, the 2800 to 3000 cm−1 region associated with carbon–hydrogen stretches, and from 3200–3700 cm−1 associated with the oxygen–hydrogen stretches of water. The relative intensities of these features showed a strong dependence on particle size. In particular, submicrometer particles exhibited a larger amount of organic matter compared to supermicrometer particles. However, for external surfaces of homogeneous SSA particles (i.e. particles without a solid inclusion), and also the interfaces of mixed-phase particles, there was a strong SFG response in the aliphatic C–H stretching region for both sub- and supermicrometer particles. This finding suggests that organic material present in supermicrometer particles primarily resides at the interface. The presence of methylene contributions in the SFG spectra indicated disordered alkyl chains, in contrast to what one might expect for a surfactant layer on a sea salt particle. Changes in peak frequencies and relative intensities in the C–H stretching region are seen for some particles after the addition of bacteria, phytoplankton, and growth medium to the seawater. This study provides new insights into the bulk and surface composition of SSA particles and represents a step forward in our understanding of this globally abundant aerosol. It also provides insights into the development of model systems for SSA that may more accurately represent the organic layer at the surface.
.jpg)


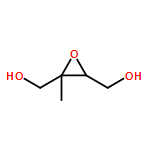




![2-Butenal, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-methyl-, (2E)-](http://img.cochemist.com/ccimg/145000/144950-82-3.png)
![2-Butenal, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-methyl-, (2E)-](http://img.cochemist.com/ccimg/145000/144950-82-3_b.png)
![2,5-Pyrrolidinedione, 1-[(1-oxo-10-undecenyl)oxy]-](http://img.cochemist.com/ccimg/110700/110661-49-9.png)
![2,5-Pyrrolidinedione, 1-[(1-oxo-10-undecenyl)oxy]-](http://img.cochemist.com/ccimg/110700/110661-49-9_b.png)


























![Bicyclo[3.1.1]hept-2-ene, 6,6-dimethyl-, (1S)-](http://img.cochemist.com/ccimg/24000/23978-82-7.png)
![Bicyclo[3.1.1]hept-2-ene, 6,6-dimethyl-, (1S)-](http://img.cochemist.com/ccimg/24000/23978-82-7_b.png)

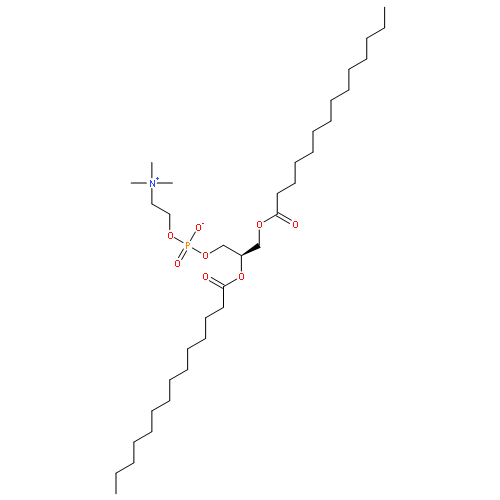












![1-Propanaminium,N,N,N-trimethyl-2,3-bis[[(9Z)-1-oxo-9-octadecen-1-yl]oxy]-](http://img.cochemist.com/ccimg/113700/113669-21-9.png)
![1-Propanaminium,N,N,N-trimethyl-2,3-bis[[(9Z)-1-oxo-9-octadecen-1-yl]oxy]-](http://img.cochemist.com/ccimg/113700/113669-21-9_b.png)


![3,5,9-Trioxa-4-phosphaheptacos-18-en-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[[(9Z)-1-oxo-9-octadecen-1-yl]oxy]-, innersalt, 4-oxide, (7R,18Z)-](http://img.cochemist.com/ccimg/4300/4235-95-4.png)
![3,5,9-Trioxa-4-phosphaheptacos-18-en-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[[(9Z)-1-oxo-9-octadecen-1-yl]oxy]-, innersalt, 4-oxide, (7R,18Z)-](http://img.cochemist.com/ccimg/4300/4235-95-4_b.png)


![2,5-Pyrrolidinedione, 1-[[1-oxo-11-(trichlorosilyl)undecyl]oxy]-](http://img.cochemist.com/ccimg/870300/870246-45-0.png)
![2,5-Pyrrolidinedione, 1-[[1-oxo-11-(trichlorosilyl)undecyl]oxy]-](http://img.cochemist.com/ccimg/870300/870246-45-0_b.png)