Co-reporter:Md Abdul Goni, Edward Rosenberg, Roberto Gobetto, Michele Chierotti
Journal of Organometallic Chemistry 2017 Volume 845(Volume 845) pp:
Publication Date(Web):15 September 2017
DOI:10.1016/j.jorganchem.2017.05.036
•Two Ru pincer complexes have been immobilized on a silica polyamine composite and are shown to be active catalysts for dehydrogenative coupling of alcohols.•The amine surface provides the required base needed to activate the complexes for the catalysis.•Model reactions studied in solution show the regiochemistry of electrophilic aromatic substitution with both para-and meta isomers being formed.•Catlyst leaching proved to be a problem but conversions could be obtained up to 5 cycles.Heterogenization of catalytically important homogeneous catalysts on solid supports has become an expanding area of research. PNN and PONOP ruthenium pincer complexes were immobilized on a silica poly(allylamine) composite, BP-1 by a two-step Mannich reaction. The complexes on BP-1 were characterized by solid state NMR, FT-IR, elemental analysis, and metal digestion studies. Model solution experiments were carried out to determine the site of electrophilic substitution on the pyridine ring of the pincer complexes and revealed substitution in both the meta- and para-position. The catalytic reactivity of immobilized (PNN)RuH(Cl)(CO) and (PONOP)RuH(Cl)(CO) on BP-1 was studied for the dehydrogenative coupling of alcohols to esters with the liberation of H2. Moderate to good ester yields were realized with both immobilized systems without using the base required for the homogeneous reaction and also in the presence of KOH. The homogeneous model reactions required a base for ester formation. The amine functionality on BP-1 served as the base to generate the active pincer catalyst on the BP-1 surface. Both immobilized catalysts were recycled for multiple alcohol reaction cycles. Four-step control experiments were carried out using an alcohol and both immobilized systems. The results revealed the heterogeneity of the alcohol catalysis by both BP-1-Ru-PNN and BP-1-Ru-PONOP systems. This study has opened a new catalytic methodology for reactions where base is required for catalyst activation, by using a solid support with basic functionality.Download high-res image (194KB)Download full-size image
Co-reporter:Shyam Pohkrel, Dan Decato, Edward Rosenberg, J.B. Alexander Ross, Michelle Terwilliger
Journal of Organometallic Chemistry 2017 Volumes 849–850(Volumes 849–850) pp:
Publication Date(Web):1 November 2017
DOI:10.1016/j.jorganchem.2017.03.026
•Two new Ru 2-ppy have been synthesized and their physical and chemical properties are reported.•A series of Ru bpy complexes are also reported the methyl groups all show virtual coupling.•The phenyl ring of the PPhMe2 shows π-π stacking interactions with phenyl rings of the bpy ligand.The synthesis, structure and photophysical properties of the complexes [Ru[(CO)(TFA) (PPh3)2(L)][(L = ppy = 2-phenylpyridine, (1a); L = 2–(p–tolyl)pyridine] (1b), are reported. The complexes were characterized by UV-VIS, IR and NMR and by single-crystal X-ray diffraction techniques. We also report the synthesis, structure and photophysical properties of [Ru(CO)(L)(PPhMe2)2(L′)]+[PF6]− [L′ = bipyridine, L = TFA, (3a); L = H, (3b) and L = H, L′ = 4,4′-dimethlyl bipyridine (3c)]. These compounds were characterized by UV-VIS, IR and NMR techniques and by a single crystal X-ray diffraction in the case of 3a. The solid state structure of [Ru(Me2PhP)2(CO)2(TFA)2 (2) which is the starting material for the synthesis 3a-3c is also reported to verify the trans relationship of the less bulky PPhMe2 and for comparison with the previously reported PPh3 analogs. The purpose of this study was to determine the impact, if any, of replacing bpy with ppy in the case of 1a and alkylation of the benzene ring in the case of 1b on the photophysical and electrochemical properties compared to related Ru(bpy) complexes. In contrast to the bpy analogs 1a and 1b showed reversible 1e− oxidations and blue-shifted MLCT absorptions. In the case of 3a-3c we were interested in the effect on the photophysical properties of substituting PPh3 with the less bulky but more electron donating PPhMe2. There were only minor changes in the photophysical and electrochemical properties relative to the previously reported PPh3 analogs.Download high-res image (159KB)Download full-size image
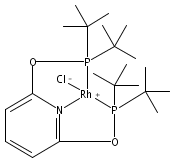
Co-reporter:Md Abdul Goni, Edward Rosenberg, Shesharao Meregude, Geoffrey Abbott
Journal of Organometallic Chemistry 2016 Volume 807() pp:1-10
Publication Date(Web):1 April 2016
DOI:10.1016/j.jorganchem.2016.01.032
•Four pincer complexes were grafted to a hybrid surface using the Mannich reaction.•The best method was reaction of the preassembled complex with the imine Mannich intermediate.•Complex loading was best analyzed by complete digestion of the loaded composite.Immobilization of catalytically active transition metal complexes on silica polyamine composite (SPC) surfaces offers many advantages for applications in catalysis particularly for catalyst recovery and reuse. We report here the immobilization of PONOP pincer complexes of Ru, Rh, Ni and Pd on the poly(allylamine) SPC, BP-1 using the Mannich reaction. Three different methods have been investigated for synthesizing the PONOP pincer transition metal complexes on BP-1: 1) direct reaction of the preformed pincer complexes using a two step Mannich reaction; 2) immobilization of the PONOP ligand using the Mannich reaction followed by the addition of a transition metal compound of a given metal; 3) the stepwise construction of PONOP on BP-1 followed by addition of a transition metal compound. The immobilized complexes on BP-1 were characterized by FT-IR, solid-state CPMAS 13C and 31P NMR, as well as elemental analysis. Anchoring of the complexes on BP-1 was also evaluated by the metal loading data obtained from the digestion of the loaded composites followed by Atomic Absorption Spectroscopy (AAS) or Inductively Coupled Plasma Atomic Emission Spectroscopy (ICPAES). The results showed that method 1 works better for the loading of pincer complexes on the SPC than methods 2 and 3. In the case of the Ru and Ni pincer complexes reasonable agreement with the phosphorous analysis was realized, while for the Pd complex values were high relative to the loading predicted from the phosphorus analysis, indicating the formation of the Pd nanoparticles on the surface during immobilization. For the Rh and Ru immobilized complexes with methods 2 & 3, metal loading was lower than the phosphorous analysis and this is attributed to entrained triphenylphosphine from the starting rhodium and ruthenium complexes based on the 13C and 31P CPMAS NMR data. Solution experiments using the PONOP pincer ligand and the Ru(PONOP) complex with n-butyl amine were conducted to model the site of electrophilic aromatic substitution on the pyridine ring. It was found that substitution both meta- and para-to the nitrogen takes place and this helped in the interpretation of the solid-state data.Three different methods of grafting a range of transition metal pincer complexes to the surface of silica polyamine composites have been studied. The best method uses the Mannich reaction and the preassembled complex.
Co-reporter:Edward Rosenberg;Geoffrey Abbott;J. B. Alexer Ross;Riley McVay ;Michelle Terwilliger
Macromolecular Symposia 2016 Volume 364( Issue 1) pp:47-55
Publication Date(Web):
DOI:10.1002/masy.201500179
Summary
Luminescent ruthenium diimine complexes have been covalently bound to the surface of a silica polyamine composite (SPC) using peptide coupling agents. The loading of the complexes using this route is quite low (∼0.01–0.04 mmol/g) leaving sufficient surface amines to coordinate added metal ions. When the composite particles containing the Ru complexes are exposed to solutions of Cu2+, Ni2+ or Zn2+, luminescence is quenched with efficiencies that follow concentration dependence and the relative binding affinities of the ions. When heavy metal ions such as mercury or lead are adsorbed onto the same surface, luminescence is enhanced by a factor of ∼3.5. When the complexes are exposed to these metals in solution, no quenching or enhancement is observed. Both phenomena were shown to be the result of adsorption of the cations onto the polyamine surface by using the Stern-Volmer relationship. The mechanism of both quenching and enhancement is discussed and the options for further development of this novel metal sensing technique are presented.
Co-reporter:Geoffrey Abbott;Robert Brooks
Journal of Applied Polymer Science 2015 Volume 132( Issue 30) pp:
Publication Date(Web):
DOI:10.1002/app.42271
ABSTRACT
The structure and properties of silica polyamine composites (SPC) made from microparticles of amorphous silica gel (300–600 microns) and silica nanoparticles (10–20 nm) modified with aminopropyltrimethoxysilane (APTMS), poly(allylamine) (PAA) or poly(ethyleneimine) (PEI) have been studied. The APTMS nano-hybrids showed batch capacities for copper equal to or better than the corresponding polymer-based micro-hybrids. Loading of the PEI on the nanoparticles was independent of molecular weight of the polymer. Dynamic light scattering measurements showed that the SiO2 nanoparticles and the composites made from them aggregate in water and the degree of aggregation is dependent on the surface modification. All of the amine-modified materials were catalysts for the Knoevenagel reaction but interestingly, the microparticles modified with APTMS were better catalysts than the corresponding nanoparticles or the polyamine modified composites. Solid-state 19Si NMR has been used to elucidate the surface structure of the various composites. © 2015 Wiley Periodicals, Inc. J. Appl. Polym. Sci. 2015, 132, 42271.
Co-reporter:Edward A. Karakhanov;Anton L. Maximov;Anna V. Zolotukhina;Nadezhda Yatmanova
Applied Organometallic Chemistry 2015 Volume 29( Issue 11) pp:777-784
Publication Date(Web):
DOI:10.1002/aoc.3367
A series of Pd–Ag mixed-metal nanocatalysts were prepared by reduction of Pd–Ag salts in the presence of poly(propylene imine) dendrimers, which were covalently bound to the surface of a silica polyamine composite, BP-1 (polyallylamine covalently bound to a silanized amorphous silica gel). Three different Pd-to-Ag ratios were evaluated (50:50, catalyst 1; 40:60, catalyst 2; 60:40, catalyst 3) with the goal of determining how the amount of Ag effects selectivity, rate and conversion in the selective reduction of alkynes, such as phenylacetylene and 1- or 4-octyne, to the corresponding alkenes. Conditions for the catalysis are reported where there is improved selectivity without a serious reduction in rate when compared with the analogous Pd-only catalysts. Catalyst 2 worked best for phenylacetylene and catalyst 3 worked best for the octynes. The catalysts could be reused seven times without loss of activity. Copyright © 2015 John Wiley & Sons, Ltd.
Co-reporter:Mauro Ravera, Ayesha Sharmin, Edward Rosenberg
Inorganica Chimica Acta 2015 Volume 429() pp:87-92
Publication Date(Web):1 April 2015
DOI:10.1016/j.ica.2015.02.005
•The electrochemical behavior of a series of Ru(II)-diimine complexes is discussed.•Strong binding to bovine serum albumin of two model complexes was observed.•Evidence for differential intercalation of the model complexes with DNA is presented.•Phosphine ligand has a strong impact on the electrochemical behavior.The electrochemical behavior of the ruthenium(II) diimine complexes [Ru(CO)(CF3CO2)(dppene)(5-R-phen)][PF6] (dppene = 1,2-diphenylphosphinoethene; phen = 1,10-phenanthroline; R = H, 1; R = NH2, 2; R = cholestoryl carbamate, 3; R = 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine, 4), [Ru(CO)(H)(4,4′-R-bpy)(R′Ph2P)2][PF6] (bpy = 2,2′-bipyridine; R = H, R′ = Ph, 5; R = H, R′ = 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine, 6; R = 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine, R′ = Ph, 7), [Ru(bpy)2(5-R-phen)][PF6]2 (R = NH2, 8; R = cholestoryl carbamate, 9; R = 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine, 10) is reported. Complexes 1–6 give cyclovoltammetric (CV) responses with multiple ill-defined reduction waves and one oxidation wave, all of which were chemically irreversible. Complexes 5 and 7, containing axially coordinating phosphines, showed reversible oxidation and reduction CV responses, while 6 showed redox waves similar to 3. Complexes 8–10 show a metal-centered irreversible oxidation around +1.4 V that, in the case of 8 and 9, is heavily modified by adsorption phenomena. In the negative part of the CV, 9 and 10 show a single chemically and electrochemically reversible 1e− reduction both at E°′ = −1.29 V, about 500 mV cathodically shifted with respect to 8. The interactions of complexes 1 and 2 with bovine serum albumin (BSA) and double stranded DNA (ds-DNA) were also studied by electrochemical methods. Both complexes showed strong binding to BSA. Evidence for intercalation of both complexes with DNA is presented, with 1 showing a stronger interaction than 2.The electrochemical behavior of a series of Ru(II)-diimine complexes is discussed. In particular, electrochemical techniques were use to evaluate the binding to bovine serum albumin of two model complexes. Moreover, evidence for differential intercalation of the model complexes with DNA is presented.
Co-reporter:Edward Karakhanov, Anton Maximov, Yulia Kardasheva, Vera Semernina, Anna Zolotukhina, Andrey Ivanov, Geoffrey Abbott, Edward Rosenberg, and Vladimir Vinokurov
ACS Applied Materials & Interfaces 2014 Volume 6(Issue 11) pp:8807
Publication Date(Web):April 25, 2014
DOI:10.1021/am501528a
New heterogeneous hydrogenation catalysts, based on Pd nanoparticles and polypropyleneimine (PPI) dendrimers of the third generation that have been covalently grafted to a silica surface modified with polyallylamine (PAA) have been synthesized. The final products were characterized by TEM, XPS, and solid-state NMR spectroscopy. The synthesized materials are effective catalysts for selective hydrogenation of dienes to monoenes and phenyl acetylene to styrene at very high substrate/Pd ratios with turnover rates higher than related Pd nanoparticle catalysts. The synthesized catalysts can be reused without any loss of activity in the case of styrene and isoprene.Keywords: amine-modified silica; dendrimers; hybrid-materials; hydrogenation; Pd catalysis;
Co-reporter:Geoffrey Abbott, Robert Brooks, Edward Rosenberg, Michelle Terwilliger, J. B. Alexander Ross, and Ogar O. L. Ichire
Organometallics 2014 Volume 33(Issue 10) pp:2467-2478
Publication Date(Web):May 8, 2014
DOI:10.1021/om401153x
Ruthenium complexes of the general formula [Ru(CO)(H)(L2)(L′2)][PF6] (L2 = trans-2PPh3, L′ = η2-4,4′-dicarboxybipyridine (1); L2 =trans-2Ph2PCH2CH2COOH, L′2 = bipyridine (2); L2 = Ph2PCHCHPPh2, L′ = η2-5-amino-1,10-phenanthroline (3); L2 = trans-2PPh3, L′2 = η2-4-carboxaldehyde-4′-methylbipyridine (4)) have been shown to have longer emission lifetimes and higher quantum yields in solution compared with more symmetrical molecules such as [Ru(bpy)3][Cl]2. Compound 4 is obtained as a mixture with the corresponding acetal, 4′. These less symmetrical complexes have been covalently immobilized on the surface of silica polyamine composites, and their photophysical properties have been studied. The surface-bound complexes have been characterized by solid-state CPMAS 13C, 31P, and 29Si NMR, UV–vis, and FT-IR spectroscopies. Excited-state lifetime studies revealed that, in general, the lifetimes of the immobilized complexes are 1.4 to 8 times longer than in solution and are dependent on particle size (300–500 μm versus 10–20 nm average diameter silica gels), polymer structure (linear poly(allylamine) versus branched poly(ethylenimine)), and the type of surface tether. One exception to this trend is the previously reported complex [Ru(bpy)2(5-amino-1,10-phenanthroline)][PF6]2 (5), where only a slight increase in lifetime is observed. Only minor changes in emission wavelength are observed for all the complexes. This opens up the possibility for enhanced heterogeneous electron transfer in photocatalytic reactions.
Co-reporter:Edward Rosenberg;Rakesh Kumar
Journal of Cluster Science 2014 Volume 25( Issue 1) pp:239-252
Publication Date(Web):2014 January
DOI:10.1007/s10876-013-0635-7
An overview of the dynamical processes involving the hydrido ligand in triosmium and triruthenium carbonyl clusters is presented. The relationship between the mechanisms of hydride motions and the other ligands in the cluster are discussed for mono- di- and trihydrido-clusters. In addition, the reactivity of the electron deficient 46e− cluster, (μ-H)(μ3-η2-C9H5N-4-CHO)Os3(CO)9 (1) with hydrogen is reported. The reaction gives two isomeric trihydrido clusters, H(μ-H)2(μ3-η2-C9H5N-4-CHO)Os3(CO)8 (2) and (μ-H)3(μ3-η2-C9H5N-4-CHO)Os3(CO)8 (2′) in low yield along with trace amounts of other hydrido clusters. Reaction of the inseparable mixture of 2 and 2′ with triphenylphosphine at ambient temperatures gives two related addition products H(μ-H)2(μ-η2-C9H5N-4-CHO)Os3(CO)8PPh3 (3) and (μ-H)3(μ-η2-C9H5N-4-CHO)Os3(CO)8PPh3 (3′) in a 5:1 ratio. These results contrast with the previously reported trihydrido-derivatives of triosmium μ3-η2-imidoyl clusters where only analogues of 2 and 3 are obtained. Clusters 2 and 2′ are rigid on the NMR time scale while 3 exhibits dynamical behavior in the temperature range of −50 to +25 °C. Cluster 3′ is stereochemically rigid in this temperature range. The dynamical behavior of 3 involves the exchange of the terminal and bridging hydrides coupled with tripodal motion of the phosphine substituted osmium atom, a process virtually identical to previously reported trihydrides of the μ3-η2-imidoyl triosmium clusters.
Co-reporter:Ayesha Sharmin, Luca Salassa, Edward Rosenberg, J. B. Alexander Ross, Geoffrey Abbott, Labe Black, Michelle Terwilliger, and Robert Brooks
Inorganic Chemistry 2013 Volume 52(Issue 19) pp:10835-10845
Publication Date(Web):September 24, 2013
DOI:10.1021/ic400706u
The luminescent, mono-diimine ruthenium complexes [(H)Ru(CO)(PPh3)2(dcbpy)][PF6] (1) (dcbpy = 4,4′-dicarboxy-2,2′-bipyridyl) and [(H)Ru(CO)(dppene)(5-amino-1,10-phen)][PF6] (2) (dppene = bis(diphenylphosphino)ethylene; phen = phenanthroline) were conjugated with 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (DPPE) and with cholesterol in the case of complex 2. Using standard conjugation techniques, compound 1 gives the bis-lipid derivative [(H)Ru(CO)(PPh3)2(dcbpy-N-DPPE2)][PF6] (3), while 2 provides the monolipid conjugate [(H)Ru(CO)(dppene)(1,10-phen-5-NHC(S)-N-DPPE)][PF6] (4) and the cholesterol derivative [(H)Ru(CO)(dppene)(1,10-phen-5-NHC(O)Ocholesteryl)][PF6] (5). These compounds were characterized by spectroscopic methods, and their photophysical properties were measured in organic solvents. The luminescence of lipid conjugates 3 and 4 is quenched in organic solvents while compound 4 shows a weak, short-lived, blue-shifted emission in aqueous solution. The cholesterol conjugate 5 shows the long-lived, microsecond-time scale emission associated with triplet metal-to-ligand charge-transfer excited states. Incorporation of conjugate 3 in lipid bilayer vesicles restores the luminescence, but with blue shifts (∼80 nm) accompanied by nanosecond-time scale lifetimes. In the vesicles conjugate 4 shows a short-lived and blue-shifted emission similar to that observed in solution but with increased intensity. Conjugation of the complex [(H)Ru(CO)(PhP2C2H4C(O)O-N-succinimidyl)2(bpy)][PF6] (6″) (bpy = 2,2′-bipyridyl) with DPPE gives the phosphine-conjugated complex [(H)Ru(CO)(PhP2C2H4C(O)-N-DPPE)2(bpy)][PF6] (7). Complex 7 also exhibits a short-lived and blue-shifted emission in solution and in vesicles as observed for complexes 3 and 4. We have also conjugated the complex [Ru(bpy)2(5-amino-1,10-phen)][PF6]2 (8) with both cholesterol (9) and DPPE (10). Neither complex 9 nor the previously reported complex 10 exhibited the blue shifts observed for complexes 3 and 4 when incorporated into large unilamellar vesicles (LUVs). The anisotropies of the emissions of complexes 3, 4,and 7 were also measured in LUVs, and those of complex 5 were measured in both glycerol and LUVs. High fundamental anisotropies were observed for complexes 3, 4, and 7.
Co-reporter:Rakesh Kumar, Edward Rosenberg, Miriam Inbar Feske, Antonio G. DiPasquale
Journal of Organometallic Chemistry 2013 734() pp: 53-60
Publication Date(Web):
DOI:10.1016/j.jorganchem.2012.11.003
Co-reporter:Jesse J. Allen, Edward Rosenberg, Erik Johnston, and Carolyn Hart
ACS Applied Materials & Interfaces 2012 Volume 4(Issue 3) pp:1573
Publication Date(Web):February 24, 2012
DOI:10.1021/am201761m
A sol–gel method has been developed for the synthesis of composite materials analogous to the previously reported and commercialized silica polyamine composite (SPC) materials made from amorphous silica. Monolithic xerogels were formed using a two-step procedure with no templating agent using acid catalyzed followed by base catalyzed hydrolysis. This reaction was followed by 1H NMR. Initial sol–gels were formed using a methyltrimethoxysilane (MTMOS) and 3-chloropropyltrimethoxysilane (CPTMOS) mixture. Elemental analyses and 13C CPMAS NMR confirmed incorporation of both monomeric units into the surface structure. Some control over surface morphology was achieved by adjusting synthetic conditions. The resulting xerogels were reacted with poly(allylamine) (PAA) to give composite materials which showed much lower metal ion capacities than the commercially available amorphous silica analogs. The low degree of reaction of the chloropropyl groups indicated they were not surface-available to the polyamine. Addition of tetramethoxysilane (TMOS) produced a structural matrix and resulted in higher chloride utilization (reaction of surface chloropropyl groups with the polyamine). The ratio of the three siloxane monomeric components was varied until the resulting polyamine composite xerogels had metal capacities comparable with the commercialized SPC materials. These composites had narrower average pore size distributions and fewer small pores. Further modification of these composites with metal selective ligands showed material characteristics similar to those of commercially available SPC materials. Subjecting a composite made by the sol–gel route to one thousand load-strip cycles with Cu2+ shows essentially no loss in metal capacity, and this robustness compares favorably with that observed for the SPC made from amorphous silica gels.Keywords: chemical synthesis; composite materials; NMR; sol−gel; surface properties;
Co-reporter:Jesse Allen;Eduard Karakhanov;Sergey V. Kardashev;Anton Maximov;Anna Zolotukhina
Applied Organometallic Chemistry 2011 Volume 25( Issue 4) pp:245-254
Publication Date(Web):
DOI:10.1002/aoc.1749
Abstract
The transition metal compounds Pd(OAc)2, RhCl3·4H2O and RuCl3 · nH2O were adsorbed onto the nanoporous silica polyamine composite (SPC) particles (150–250 µm), WP-1 [poly(ethyleneimine) on amorphous silica], BP-1 [poly(allylamine) on amorphous silica], WP-2 (WP-1 modified with chloroacetic acid) and BP-2 (BP-1 modified with chloroacetic acid). Inductively coupled plasma-atomic emission spectrometry analysis of the dried samples after digestion indicated metal loadings of 0.4–1.2 mmol g−1 except for RhCl3·4H2O on BP-2 which showed a metal loading of only 0.1 mmol g−1. The metal loaded composites were then screened as hydrogenation catalysts for the reduction of 1-octene, 1-decene, 1-hexene and 1, 3-cyclohexadiene at a hydrogen pressure of 5 atm in the temperature range of 50–90 °C. All 12 combinations of SPC and transition metal compound proved active for the reduction of the terminal olefins, but isomerization to internal alkenes was competitive in all cases. Under these conditions, selective hydrogenation of 1,3-cyclohexadiene to cyclohexene was observed with some of the catalysts. Turnover frequencies were estimated for the hydrogenation reactions based on the metal loading and were in some cases comparable to more conventional heterogeneous hydrogenation catalysts. Examination of the catalysts before and after reaction with X-ray photoelectron spectroscopy and transmission electron microscopy revealed that, in the cases of Pd(OAc)2 on WP-2, BP-1 and BP-2, conversion of the surface-ligand bound metal ions to metal nano-particles occurs. This was not the case for Pd(OAc)2 on WP-1 or for RuCl3 · nH2O and RhCl3· 4H2O on all four composites. The overall results are discussed in terms of differences in metal ion coordination modes for the composite transition-metal combinations. Suggested ligand interactions are supported by solid state CPMAS 13C NMR analyses and by analogy with previous structural investigations of metal binding modes on these composite materials. Copyright © 2011 John Wiley & Sons, Ltd.
Co-reporter:Jesse Allen, Matthew Berlin, Mark Hughes, Erik Johnston, Varadharajan Kailasam, Edward Rosenberg, Tova Sardot, Jessica Wood, Carolyn Hart
Materials Chemistry and Physics 2011 Volume 126(Issue 3) pp:973-982
Publication Date(Web):15 April 2011
DOI:10.1016/j.matchemphys.2010.11.053
The factors affecting the rate of silica leaching in alkaline aqueous media from surface silanized, nanoporous, amorphous, silica gels and from silanized silica gels that have been modified with polyamines to form the previously reported silica polyamine composites (SPCs), BP-1 and BP-2 have been investigated. Silanization with alkyl trichlorosilanes slows the rate of silica leaching relative to the unmodified silica gels. The use of bulkier aryl silanes somewhat decreases the silica leaching under the same conditions. Interestingly, after modification of the silanized silica with poly(allylamine) (PAA) to make BP-1, the leaching increases, but subsequent modification of the SPC with chloroacetic acid to make BP-2, quenches this increase. A mechanism explaining these results is discussed. Analogous composites have been prepared using sol–gel chemistry. These materials were characterized and their silica leaching properties were compared with the original BP-1. CPMAS 13C and 29Si NMR of the various surfaces have been applied to better understand the nature of the modified surfaces. Significant changes in the nature of the surface siloxanes are observed for the different matrices and on their conversion to the polyamine composite. Scanning electron microscopy and pore size distributions for the composites made from commercial silica gel and from sol–gel chemistry are also reported and compared.Research highlights▶ Silica leaching rates for silica polyamine composites have been measured. ▶ The surface features of the composites with different silane anchors have been evaluated. ▶ The leaching characteristics of the composites are compared with similar materials made by the sol–gel method. ▶ Solid state NMR techniques, porosimetry and electron microscopy have been used to characterize all the materials discussed.
Co-reporter:Matt Berlin;Jesse Allen;Varadharajan Kailasam;David Rosenberg
Applied Organometallic Chemistry 2011 Volume 25( Issue 7) pp:530-536
Publication Date(Web):
DOI:10.1002/aoc.1798
Abstract
Rice hull ash (RHA) was converted to amorphous silica gel using a modified version of published literature procedures. The gels were characterized by a comparison of their Cross Polarization Magic Angle Spinning 29Si NMR and scanning electron microscopy images with commercial silica gels. The resulting gels were silanized with a 7.5:1 mixture of methyltrichlorosilane and chloropropyltrichlorosilane and then reacted with poly(allylamine) (PAA) to produce the silica polyamine composite (SPC) BP-1. The BP-1 was then further modified with pyridine-2-carboxaldehyde to form the copper-selective SPC, CuSelect. This procedure follows that used to produce the commercialized version of these composite materials from commercially available amorphous silica gels. The composites were characterized by solid-state NMR techniques, elemental analysis, SEM, porosimetry and metal ion capacity and selectivity. The overall goal of the project was to determine the feasibility of using RHA to make SPC. The observed strengths and weaknesses of this approach are discussed. Copyright © 2011 John Wiley & Sons, Ltd.
Co-reporter:Jesse Allen, Edward Rosenberg, Michele R. Chierotti, Roberto Gobetto
Inorganica Chimica Acta 2010 Volume 363(Issue 3) pp:617-624
Publication Date(Web):15 February 2010
DOI:10.1016/j.ica.2008.12.031
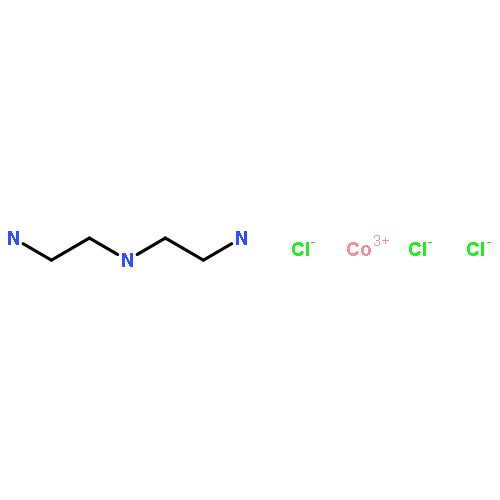
Coordination of CoCl2 solutions to the silica polyamine composite, WP-1, made with the branched polymer polyethylenimine (PEI) shows irreversible binding resulting from surface oxidation of the Co2+–Co3+. This is not the case for the silica polyamine composite BP-1 made with the linear polymer polyallylamine where reversible binding and no oxidation is observed. These observations suggested that oxidation of the cobalt was related to the greater coordination number available with the branched polyamine relative to the linear polyamine. A study of the kinetics of cobalt binding to WP-1 indicated initial loading of Co2+ at relatively low coordination number followed by desorption of Co2+ leading to oxidation and irreversible binding. Exclusion of oxygen from the composite-cobalt solution mixtures resulted in irreversible binding at a level that was 14% of the initial experiments. These observations prompted us to undertake a study to elucidate the coordination number around cobalt in the case of the branched polymer PEI. Towards this end, we have synthesized the model complexes [(tren)Co(H2O)2]3+3Cl− (tren = tris(2,2′,2″aminoethyl)amine) and [(dien)Co(H2O)3]3+3Cl− (dien = diethylenetriamine). The UV–Vis spectra of these model complexes were compared with Co3+ coordinated to PEI in solution and it was concluded that the UV–Vis spectrum of the tren complex was closer to that observed for the solution UV–Vis spectrum of Co3+–PEI. These data indicated that coordination of four amines was needed to drive surface oxidation under ambient conditions. In order to further elucidate the coordination number of a metal coordinated to the surfaces of WP-1 and BP-1, we reacted these composites with the probe molecule [Ru(CO)3(TFA)3]−K+ (TFA = trifluoroacetate) (1) where the carbonyl stretching frequencies could be used as a measure of coordination number and geometry of the adsorbed complex. The IR of this complex on WP-1 indicated a monocarbonyl species while the IR of this complex on BP-1 indicated the presence of dicarbonyl species on the surface. These data are consistent with a coordination number of four amines in the case of WP-1 and the coordination of two amines in the case of BP-1 based on our previous studies of the solution coordination chemistry of the 1. Subsequent 13C CPMAS solid-state NMR on 13CO enriched samples of the 1 adsorbed onto BP-1 and WP-1 were consistent with the IR data.Cobalt(II) adsorbed onto a branched polyamine, grafted onto amorphous silica spontaneously oxidizes in the presence of oxygen to Co(III). When adsorbed onto the silica polyamine composite made with a linear polyamine this does not occur. Spectroscopic studies using model cobalt complexes and a reactive ruthenium probe molecule indicate that octahedral metal centers bind to four amines in the case of the branched polymer and to two amines in the case of the linear polymer.
Co-reporter:Yuen Onn Wong;Paul Mira
Journal of Applied Polymer Science 2010 Volume 115( Issue 5) pp:2855-2864
Publication Date(Web):
DOI:10.1002/app.31229
Abstract
A series of oxine ligands, 5-X, 8OHC9H6N (X = H, Cl, SO3H) have been covalently bound to a silica gel polyamine composite made from a silanized amorphous silica xerogel and poly(allylamine) (BP-1) by the Mannich reaction. The resulting modified composites WP-4(X = H), CB-1(X = Cl), and SB-1(X = SO3H) were characterized by elemental analysis, FTIR, and solid state CPMAS-13C-NMR. Using the analytical data before and after the ligand modification, the ligand loading could be estimated and in combination with metal ion capacities a metal to ligand ratio could be obtained. The composites WP-4 and CB-1 both showed ratios of approximately 1 : 1 while the sulfonate modified composite, SB-1, showed a metal to ligand ratio of 1 : 2. This is tentatively interpreted in terms of a zwitterionic oxine as the dominant species at pH = 2 where the sulfonic acid group is ionized, the quinoline nitrogen is protonated and where two sulfonate groups on adjacent oxines bind a trivalent or divalent ion. All the modified composites show a clear selectivity for trivalent over divalent ions and a good selectivity for gallium over aluminum. The substituent groups on the oxine ligand have only a minor influence on these selectivity trends but SB-1 shows slightly better capture kinetics. The selectivity for gallium over aluminum is applied to the separation of gallium from aluminum, ferrous, and zinc in an acid ore leach of solid tailings obtained from a zinc mine using WP-4. © 2009 Wiley Periodicals, Inc. J Appl Polym Sci, 2010
Co-reporter:Varadharajan Kailasam, Edward Rosenberg and Daniel Nielsen
Industrial & Engineering Chemistry Research 2009 Volume 48(Issue 8) pp:3991
Publication Date(Web):March 23, 2009
DOI:10.1021/ie8016362
Silica polyamine composites (SPC) made from silanized amorphous nanoporous silica gel and poly(allylamine) (BP-1) were functionalized with phosphorus acid using the Mannich reaction, resulting in a phosphonic acid modified composite (BPAP). Successful immobilization of the phosphonic acid ligand was confirmed by mass gain, elemental analysis, IR, and solid-state NMR. The modified composite had a ligand loading of 1.5 mmol/g, corresponding to N/P ratio of 0.73. Zirconium(IV) was immobilized on BPAP with a loading of 1.12 mmol/g. Zirconium loading was analyzed by mass gain, ICP/AES, and SEM/EDX. Arsenate anions strongly adsorbed on the ZrBPAP composite in the pH range 2−8, while arsenite only adsorbed well at pH 10. The sorption mechanism is a chelation between arsenate or arsenite and Zr(IV)−phosphonic acid complex of BPAP. Adsorption isotherm data were found to be well modeled by the Langmuir equation for As(V) at pH 4 with Kads = 0.016 L/g and Qm = 98 mg/g; and at pH 6 with Kads = 0.018 L/g and Qm = 56 mg/g. Regeneration of the resin was carried out successfully for As(V) and As(III) using 2M-H2SO4. Four adsorption/desorption cycles were performed for As(V) at pH 4 without significant decrease in the uptake performance. ZrBPAP capture capacity and kinetics for arsenate were tested for longevity over 1000 cycles with only a marginal loss of performance. This composite is highly selective for arsenate over sulfate (As/SO42− = 50/1) and selenate (As/Se = 20/1); lower selectivity was observed with Fe(III) and Th(IV) loaded BPAP. The significance of the observed selectivity is discussed in terms of the chemical properties of the anion and the nature of the interactions with the immobilized metal site.
Co-reporter:Ayesha Sharmin, Reuben C. Darlington, Kenneth I. Hardcastle, Mauro Ravera, Edward Rosenberg, J.B. Alexander Ross
Journal of Organometallic Chemistry 2009 694(6) pp: 988-1000
Publication Date(Web):
DOI:10.1016/j.jorganchem.2008.11.048
Co-reporter:Mark A. Hughes, Jessica Wood and Edward Rosenberg
Industrial & Engineering Chemistry Research 2008 Volume 47(Issue 17) pp:6765
Publication Date(Web):July 23, 2008
DOI:10.1021/ie800359k
Previously, silica polyamine composite (SPC) materials (BP-1 and WP-1) containing amine chelating groups were prepared using two polyamines, polyallylamine (PAA), and polyethyleneimine (PEI). In this paper the amine ligands of BP-1 and WP-1 were modified with chloroacetate, yielding the new SPC materials BP-2 and WP-2. We have found that the acetate groups were bound to the amine groups to a greater extent on the SPC prepared using PAA (BP-1) relative to the SPC prepared with PEI (WP-1). BP-2 was selective for Cu2+ over other divalent metal ions from polymetallic solutions in the pH range of 1 to 3. In contrast, WP-2 was selective for Cu2+ over other divalent metal ions at pH 1 only and coloaded significant amounts of Ni2+ at pH 2 and pH 3. Thus, polyamine structure impacts the metal selectivity of the resultant SPC materials, BP-2 and WP-2. Two novel SPCs were prepared from nitriloacetic acid (NTA) anhydride using BP-1 and WP-1, yielding BP-NT and WP-NT, respectively. The resultant materials possess a unique chelating ligand, in which an iminodiacetic acid (IDA) group is covalently bonded to the SPC amine groups via an amide bond. The two materials (BP-NT, WP-NT) have similar metal selectivity profiles indicating that polyamine structure is not influential. Increased ligand denticity and the amide linker prevent the polymer from playing a large role in selectivity. The resulting materials have the ability to remove divalent and trivalent metal ions from low pH aqueous solutions. These materials can be regenerated by treatment with acid solution and showed no evidence of amide bond hydrolysis under acidic conditions.
Co-reporter:Ayesha Sharmin, Agnese Minazzo, Luca Salassa, Edward Rosenberg, J.B. Alexander Ross, Shariff E. Kabir, Kenneth I. Hardcastle
Inorganica Chimica Acta 2008 Volume 361(Issue 6) pp:1624-1633
Publication Date(Web):5 May 2008
DOI:10.1016/j.ica.2007.03.059
The reactions of 2-amino-anthracene with [Os3(CO)10(CH3CN)2] have been studied and the products structurally characterized by spectroscopic, X-ray diffraction, photophysical and electrochemical techniques. At room temperature in CH2Cl2 two major, isomeric products are obtained [Os3(CO)10(μ-η2-(N-C(1))-NH2C14H8)(μ-H)] (1, 14%) and [Os3(CO)10(μ-η2-(N-C(3))-NHC14H9)(μ-H)] (2, 35%) along with a trace amount of the dihydrido complex [Os3(CO)9(μ-η2-(N-C(3))-NHC14H8)(μ-H)2] (3). In refluxing tetrahydrofuran only complexes 2 and 3 are obtained in 24% and 28%, respectively. A separate experiment shows that complex 1 slowly converts to 2 and that the rearrangement is catalyzed by adventitious water and involves proton transfer to the anthracene ring. Complex 1 is stereochemically non-rigid; exhibiting edge to edge hydride migration while 2 is stereochemically rigid. Complex 3 is also stereochemically non-rigid showing a site exchange process of the magnetically nonequivalent hydrides typical for trinuclear dihydrides. Interestingly, 2 decarbonylates cleanly to the electronically unsaturated 46e− cluster [Os3(CO)9(μ3-η2-(N-C(3))-NHC10H9)(μ-H)] (4, 68%) in refluxing cyclohexane, while photolysis of 2 in CH2Cl2 yields only a small amount of 3 along with considerable decomposition. The mechanism of the conversion of 1 to 2 and the dependence of the product distribution on solvent are discussed. All four compounds are luminescent with compounds 1–3 showing emissions that can be assigned to radiative decay associated with the anthracene ligand. Complexes 1–3 all show irreversible 1e− reductions in the range of −1.85–2.14 V while 4 shows a nicely reversible 1e− wave at −1.16 V and a quasi-reversible second 1e− wave at −1.62 V. Irreversible oxidations are observed in the range from +0.35 to +0.49 V. The relationship between the cluster ligand configurations and the observed electrochemical and photochemical behavior is discussed and compared with that of the free ligand.The reaction of 2-aminoanthracene with [Os3(CO)10(CH3CN)2] results in two competitive C–H activations at C(1) and C(3) resulting in two distinct reaction pathways, one of which results in an unusual proton transfer to the amine substituted aromatic ring catalyzed by water.
Co-reporter:Noorjahan Begum, Md. Iqbal Hyder, Mohammad R. Hassan, Shariff E. Kabir, Dennis W. Bennett, Daniel T. Haworth, Tasneem A. Siddiquee, Dalia Rokhsana, Ayesha Sharmin and Edward Rosenberg
Organometallics 2008 Volume 27(Issue 7) pp:1550-1560
Publication Date(Web):March 5, 2008
DOI:10.1021/om701042s
Reactions of [Ru3(CO)10(μ-dppm)] (1) and its ortho-metalated derivative [Ru3(CO)9{μ3-η3-P(C6H5)CH2P(C6H5)(C6H4)}] (11) with PhEEPh (E = Te, Se, S) have been investigated. Treatment of 1 with PhTeTePh at room temperature afforded the dinuclear compound [Ru2(CO)4(μ-TePh)2(μ-dppm)] (2) and the 54-electron triruthenium compounds [Ru3(CO)6(μ3-Te)2(μ-TePh)2(μ-dppm)] (3) and [Ru3(CO)6(μ3-Te)(μ-TePh)3(η1-COPh)(μ-dppm)] (4). Analogous reactions of 1 with PhEEPh (E = Se, S) led to [Ru2(CO)4(μ-EP[Ru3(CO)6(μ3-E)2(μ-EPh)2(μ-dppm)])2(μ-dppm)] (E = Se, 5; E = S, 8) and the 54-electron triruthenium compounds (E = Se, 6; E = S, 9), and [Ru3(CO)6(μ3-E)(μ-EPh)3(Ph)(μ-dppm)] (E = Se, 7; E = S, 10). Reactions of the ortho-metalated complex 11 with PhEEPh (E = Te, Se, S) in refluxing THF gave exclusively [Ru3(CO)6(μ-EPh)2{μ3-η3-P(C6H5)CH2P(C6H5)(C6H4)}] (E = Te, 12; E = Se, 13; E = S, 14). The new compounds have been characterized by a combination of analytical and spectroscopic methods, and molecular structures of 2−4, 7, 10, and 13 have been determined by single-crystal X-ray diffraction studies. Compounds 2, 5, and 8 have the classical “sawhorse” structure with two bridging EPh (E = Te, Se, S) moieties and one bridging dppm ligand. Compounds 3, 6, and 9 contain a Ru3 framework with two bridging EPh (E = Te, Se, S) groups, one bridging dppm ligand, and two capping chalcogenido ligands. Compound 4 contains an Ru3 core with a capping tellurido ligand, three bridging TePh moieties, one bridging dppm ligand, and a terminally coordinated benzoyl group, formed from multiple fragmentation of the PhTeTePh ligand and migratory insertion of a Ph group into a CO ligand. Compounds 7 and 10 comprise a capping chalcogenido ligand, three bridging EPh (E = Se, 7; E = S, 10) moieties, a bridging dppm ligand, and a terminally coordinated σ-bonded phenyl group. In compounds 12–14, the coordination of ortho-metalated diphosphine ligand is the same as in 11 and both the EPh moieties bridge the same unbridged Ru−Ru edge. Compounds 3, 6, 7, and 10 exhibit restricted fluxional behavior involving the μ-EPh moieties.
Co-reporter:Avthandil A. Koridze, Aleksey M. Sheloumov, Fedor M. Dolgushin, Mariam G. Ezernitskaya, Edward Rosenberg, Ayesha Sharmin and Mauro Ravera
Organometallics 2008 Volume 27(Issue 23) pp:6163-6169
Publication Date(Web):November 6, 2008
DOI:10.1021/om800589g
The digold-tetrarhenium cluster Re4(AuPPh3)2(CO)12(μ3-C≡CFc)2 (1) was obtained by the thermolysis of known complex Re2(AuPPh3)(CO)8(μ-C≡CFc) (2) and fully characterized by a single crystal X-ray diffraction study. Cluster 1 has a butterfly Re4 geometry, and both Re3 wings are capped with AuPPh3 groups. Two ferrocenylethynyl ligands symmetrically coordinated within cavity of the wings. The dihedral angle between the Re3 wings is rather narrow, 73.2°, whereas the 64e butterfly clusters usually have a more flattened structure. An examination of the carbonyl region of the 13C NMR of 1 and 2 revealed that 1 is stereochemically rigid while 2 is fluxional with respect to a σ-π interconversion of the ferrocenyl ethynyl group across the Re−Re edge of the Re2Au triangle. Complex 1 revealed a very rare electronic communication between the ferrocene units across the cluster core. According to the cyclic voltammetric (CV) data, cluster 1 undergoes two reversible redox process with ferrocene oxidations separated by 109 mV. Electronic communication between two ferrocene units of 1 is discussed on the basis of CV and NIR data.
Co-reporter:Arun K. Raha;Mohammad R. Hassan;Shariff E. Kabir
Journal of Cluster Science 2008 Volume 19( Issue 1) pp:47-62
Publication Date(Web):2008 March
DOI:10.1007/s10876-007-0158-1
Reaction of [Os3(CO)10(CH3CN)2] with thianthrene at 80 °C leads to the nonacarbonyl dihydride compound [Os3(CO)9(μ-3,4-η2-C12H6S2)(μ-H)2] (1) and the 46-electron monohydride compound [Os3(CO)9(μ3-η2-C12H7S2)(μ-H)] (2). Compound 2 reacts reversibly with CO to give the CO adduct [Os3(CO)10(μ-η2-C12H7S2)(μ-H)] (3) whereas with PPh3 it gives the addition product [Os3(CO)9)(PPh3)(μ-η2-C12H7S2)(μ-H)] (4) as well as the substitution product 1,2-[Os3(CO)10((PPh3)2] (5) Compound 2 represents a unique example of an electron-deficient triosmium cluster in which the thianthrene ring is bound to cluster by coordination of the sulfur lone pair and a three-center-two-electron bond with the C(2) carbon which bridges the same edge of the triangle as the hydride. Electrochemical and DFT studies which elucidate the electronic properties of 2 are reported.
Co-reporter:Mark Hughes;Roberto Gobetto;Dan Nielsen;Sarah Burton;Paul Mira;Alessra Viale and
Macromolecular Symposia 2006 Volume 235(Issue 1) pp:161-178
Publication Date(Web):28 MAR 2006
DOI:10.1002/masy.200650320
Summary: The surface coverage of amorphous silica gels used in the synthesis of silica polyamine composites has been investigated by 29Si NMR. By diluting the polyamine anchor silane, chloropropyl trichlorosilane, with methyl trichlorosilane it was found that surface coverage could be markedly improved for a range of amine polymers after grafting to the silica surface. The commensurate decrease in the number of anchor points and increase in the number of free amines results in an increase in metal capacity and/or an improvement in capture kinetics. Solid state CPMAS-13C NMR has been employed to investigate the structure and metal ion binding of a series of these composite materials. It is reported that the highly branched polymer, poly(ethyleneimine) (PEI) exhibits much broader 13C NMR resonances than the linear polymers poly(allylamine) (PAA) and poly(vinylamine) (PVA). These results are understood in terms of the low energy conformations calculated from molecular modeling studies. Three new applications of the technology are also presented: 1) separation of lanthanides as a group from ferric ion and all other divalent ions; 2) a multi step process for recovering and concentrating the valuable metals in acid mine drainage; 3) a process for removing low level arsenic and selenium in the presence of sulfate using immobilized cations on the composite materials.
Co-reporter:Shariff E. Kabir, Md. Arzu Miah, Nitai C. Sarker, G.M. Golzar Hossain, Kenneth I. Hardcastle, Dalia Rokhsana, Edward Rosenberg
Journal of Organometallic Chemistry 2005 Volume 690(Issue 12) pp:3044-3053
Publication Date(Web):15 June 2005
DOI:10.1016/j.jorganchem.2005.03.041
Treatment of the electronically unsaturated cluster [(μ-H)Os3(CO)8(Ph2PCH2P(Ph)C6H4)] (1) with HCl gas at ambient temperature in dichloromethane afforded (μ-H)Os3(CO)8(μ-Cl)(μ-dppm) (2) and [(μ-H)Os3(CO)7(η1-Cl)(μ-Cl)2(μ-dppm)] (3). Thermolysis of 2 at 110 °C led to an isomer of 2, 4. A similar reaction of 1 with HBr gas gave [(μ-H)Os3(CO)8(μ-Br)(μ-dppm)] (5) as the only product which does not isomerize at 110 °C. In sharp contrast, treatment of 1 with HF gas gave the protonated species [(μ-H)2Os3(CO)8(Ph2PCH2P(Ph)C6H4)]+ (6). Treatment of 1 with CF3CO2H also gave cation 6 whereas CH3CO2H yielded [(μ-H)Os3(CO)8(μ-η2-CH3CO2)(μ-dppm)] (7). Structures of 2, 3, 4 and 6 were established crystallographically. In 2, both the chloride and the hydride ligands simultaneously bridge the same Os–Os edge and the dppm spans another Os–Os bond whereas in 4, all the three ligands bridge the same Os–Os edge. Compound 3 is comprised of an open Os3 arrangement in which one chloride bridges the open Os⋯Os edge, another chloride and a hydride mutually bridge an Os–Os bond and the third chloride is terminally coordinated to one of the Os atoms of the dppm bridged edge. The cation 6 consists of a triangle of osmium atoms in which the shortest Os–Os edge is bridged by a hydride and the metallated phenyl ring and the longest edge is bridged by another hydride and the diphosphine ligand.Treatment of the electronically unsaturated cluster [(μ-H)Os3(CO)8(Ph2PCH2P(Ph)C6H4)] (1) with HCl gas at ambient temperature afforded [(μ-H)Os3(CO)8(μ-Cl)(μ-dppm)] (2) and [(μ-H)Os3(CO)7(η1-Cl)(μ-Cl)2(μ-dppm)] (3). Thermolysis of 2 at 110 °C led to isomerization of 2 to 4. A similar reaction of 1 with HBr gas gave only [(μ-H)Os3(CO)8(μ-Br)(μ-dppm)] (5) which does not isomerizes at 110 °C. In sharp contrast, 1 reacts with HF gas to give the protonated species [(μ-H)2Os3(CO)8(Ph2PCH2P(Ph)C6H4)]+ (6). Treatment of 1 with CF3CO2H also gave cation 6 whereas CH3CO2H yielded [(μ-H)Os3(CO)8(μ-η2-CH3CO2)(μ-dppm)] (7).
.jpg)
![(2,5-DIOXOPYRROLIDIN-1-YL) 2-[4-(2,5-DIOXOPYRROLIDIN-1-YL)OXYCARBONYLPYRIDIN-2-YL]PYRIDINE-4-CARBOXYLATE](http://img.cochemist.com/ccimg/116600/116565-70-9.png)
![(2,5-DIOXOPYRROLIDIN-1-YL) 2-[4-(2,5-DIOXOPYRROLIDIN-1-YL)OXYCARBONYLPYRIDIN-2-YL]PYRIDINE-4-CARBOXYLATE](http://img.cochemist.com/ccimg/116600/116565-70-9_b.png)