Co-reporter:Yunxiang Lu, Shaoze Zhang, Changjun Peng, and Honglai Liu
The Journal of Physical Chemistry C November 9, 2017 Volume 121(Issue 44) pp:24707-24707
Publication Date(Web):October 24, 2017
DOI:10.1021/acs.jpcc.7b09325
The interplay between halogen bonding and hydrogen bonding has caused recent interest in the formation of 2D self-assembled molecular arrays on solid surfaces. Herein we present a first-principles density functional theory study of the self-assemblies of two brominated anthraquione molecules on Au(111) and Au(110). The possible binding sites of these two compounds on the facets were first explored, and then various self-assembled patterns involving different halogen and hydrogen bonds were examined both in the gas phase and on the gold surface. To visually investigate the nature of lateral adsorbate–adsorbate and vertical adsorbate–substrate interactions, the atoms in molecules, noncovalent interaction index, and electron density difference analyses were undertaken. The molecules tend to be self-assembled by means of triangular binding motifs with simultaneous halogen and hydrogen bonds. Upon the formation of the dimers in the gas phase as well as on the gold surface, significant band shifts around the Fermi energy take place, due to intermolecular orbital hybridization. The simulated scan tunneling microscopy images via the Tersoff–Hamann approach are in good agreement with those determined experimentally. The results reported in this work should be of fundamental importance in the simultaneous application of these two parallel noncovalent interactions in molecular self-assembly on surfaces.
Journal of Chemical Information and Modeling January 27, 2014 Volume 54(Issue 1) pp:
Publication Date(Web):December 28, 2013
DOI:10.1021/ci400539q
Halogen bond has attracted a great deal of attention in the past years for hit-to-lead-to-candidate optimization aiming at improving drug-target binding affinity. In general, heavy organohalogens (i.e., organochlorines, organobromines, and organoiodines) are capable of forming halogen bonds while organofluorines are not. In order to explore the possible roles that halogen bonds could play beyond improving binding affinity, we performed a detailed database survey and quantum chemistry calculation with close attention paid to (1) the change of the ratio of heavy organohalogens to organofluorines along the drug discovery and development process and (2) the halogen bonds between organohalogens and nonbiopolymers or nontarget biopolymers. Our database survey revealed that (1) an obviously increasing trend of the ratio of heavy organohalogens to organofluorines was observed along the drug discovery and development process, illustrating that more organofluorines are worn and eliminated than heavy organohalogens during the process, suggesting that heavy halogens with the capability of forming halogen bonds should have priority for lead optimization; and (2) more than 16% of the halogen bonds in PDB are formed between organohalogens and water, and nearly 20% of the halogen bonds are formed with the proteins that are involved in the ADME/T process. Our QM/MM calculations validated the contribution of the halogen bond to the binding between organohalogens and plasma transport proteins. Thus, halogen bonds could play roles not only in improving drug–target binding affinity but also in tuning ADME/T property. Therefore, we suggest that albeit halogenation is a valuable approach for improving ligand bioactivity, more attention should be paid in the future to the application of the halogen bond for ligand ADME/T property optimization.
Co-reporter:Shaoze Zhang, Yunxiang LuYuchen Zhang, Changjun Peng, Honglai Liu
The Journal of Physical Chemistry C 2017 Volume 121(Issue 8) pp:
Publication Date(Web):February 9, 2017
DOI:10.1021/acs.jpcc.6b13060
The formation of halogen-bond-based 2D supramolecular assemblies on solid surfaces has become a hot research topic in recent years. Herein, we report a theoretical study of the halogen-bonded network formation of pyridine derivatives and aryl–halide molecules on graphene surface using first-principles density functional theory (DFT) calculations. To unravel the characteristics of molecule–molecule and molecule–substrate interactions, the noncovalent interaction index (NCI), electron density difference (EDD), density of state (DOS), and topographical scan tunneling microscopy (STM) image analyses were undertaken. The N···I interactions between terminal pyridyl groups and iodoperfluorobenzenes are predicted to be somewhat stronger than the molecule–surface stacking interactions and appear to be the primary interactions in the self-assembled networks, with geometries in good agreement with the experimental STM images. The results reported in this work should be of great importance in the applications of these interactions in molecular self-assembly on surfaces.
Computational and Theoretical Chemistry 2017 Volume 1115(Volume 1115) pp:
Publication Date(Web):1 September 2017
DOI:10.1016/j.comptc.2017.06.021
•Intramolecular S⋯O interactions exhibit a good linearity in crystals.•These intramolecular bonds are weak electrostatic interactions.•Intramolecular chalcogen interactions play a conformation locking role.According to our search of the Cambridge Structural Database, a huge number of crystal structures involving intramolecular CS⋯OS(C) interactions were extracted. The largest proportion of these interactions in the crystals shows rather good linearity and short interatomic S⋯O distances. Then, intramolecular CS⋯OS(C) interactions in three representative organic molecules, i.e. sulfamethizole, thioindirubin, and thiazolo-pyridinium methide, were investigated using density functional theory calculations at the M06-2x level. The effects of different substituents and intermolecular hydrogen bonds on these interactions were also examined. The presence of both electron-withdrawing and electron-donating substituents into the donor moiety tends to strengthen intramolecular S⋯O bonds, while the formation of intermolecular hydrogen bonds has a complex influence. To gain a deeper understanding of the nature and strength of these interactions, the analyses of atom in molecules, noncovalent interaction index, and natural bond orbital were undertaken. Intramolecular chalogen bonds are weak electrostatic interactions and play a key role in controlling the crystal forms of relevant organic compounds, frequently supported by intermolecular hydrogen bonds.Download high-res image (114KB)Download full-size image
Co-reporter:Shaoze Zhang, Guimin Wang, Yunxiang Lu, Weiliang Zhu, Changjun Peng, and Honglai Liu
The Journal of Physical Chemistry A 2016 Volume 120(Issue 30) pp:6089-6102
Publication Date(Web):July 18, 2016
DOI:10.1021/acs.jpca.6b05770
In this work, the interactions between imidazolium-based ionic liquids and some stable radicals based on 2,2,6,6-tetramethylpiperidine-1-yloxyl (TEMPO) have been systematically investigated using density functional theory calculations at the level of M06-2x. Several different substitutions, such as hydrogen bonding formation substituent (OH) and ionic substituents (N(CH3)3+ and OSO3–), are presented at the 4-position of the spin probe, which leads to additional hydrogen bonds or ionic interactions between these substitutions and ionic liquids. The interactions in the systems of the radicals containing ionic substitutions with ionic liquids are predicted much stronger than those in the systems of neutral radicals, resulting in a significant reduction of the mobility of ionic radicals in ionic liquids. To further understand the nature of these interactions, the natural bond order, atoms in molecules, noncovalent interaction index, electron density difference, energy decomposition analysis, and charge decomposition analysis schemes were employed. The additional ionic interactions between ionic radicals and counterions in ionic liquids are dominantly contributed from the electrostatic term, while the orbital interaction plays a major role in other interactions. The results reported herein are important to understand radical processes in ionic liquids and will be very useful in the design of task-specific ionic liquids to make the processes more efficient.
Intramolecular halogen bonds have been the subject of several current experimental and theoretical studies. In this work, intramolecular halogen bonds in a series of 1,2-aryldiyne molecules were investigated using density functional theory calculations at the M06-2x level of theory. For comparison, some dimeric complexes between halogenated aryldiynes and quinolinyl compounds were also considered. The calculated interatomic distances and interaction angles of intramolecular halogen bonds compare fairly well with those determined experimentally, and the triangle motifs retain almost perfectly planar in all the studied molecules. Many of the well-known properties of conventional halogen bonds are reproduced in intramolecular halogen bonds: the interaction strength tends to increase with the enlargement of the atomic radius of halogens (I > Br > Cl); the attachment of electron-withdrawing moieties to halogens leads to much stronger intramolecular halogen bonds; the X···N (quinolinyl) interactions are stronger than the X···O (carbonyl) halogen bonds. On the basis of the shorter interatomic distances and the larger values of electron densities at the bond critical points, intramolecular halogen bonds become stronger in strength than corresponding intermolecular halogen bonds. However, these interactions have similar structural, energetic, atoms in molecules (AIM), and noncovalent interaction index (NCI) characteristics to traditional halogen bonds. Therefore, these interactions can be recognized as halogen bonds that are primarily electrostatic in nature. Particularly, the formation of intramolecular halogen bonds gives rise to the essential coplanarity of the molecules, whereas the two subunits in the dimeric complexes deviate from planarity to a large degree. In addition, a small number of crystal structures containing intramolecular halogen bonds were retrieved from the Cambridge Structural Database (CSD), to provide more insights into these interactions in crystals. This work not only will extend the knowledge of noncovalent interactions involving halogens as electrophilic centers but also could be very useful in molecular design and synthetic chemistry.
Co-reporter:Hairong Ding, Yunxiang Lu, Yaoming Xie, Honglai Liu, and Henry F. Schaefer III
Journal of Chemical Theory and Computation 2015 Volume 11(Issue 3) pp:940-949
Publication Date(Web):February 12, 2015
DOI:10.1021/ct501020h
The M4(CO)12 molecules Co4(CO)12, Rh4(CO)12, and Ir4(CO)12 have two low-lying structures, the all-terminal structure with Td symmetry and the triply bridged structure with C3v symmetry. A total of 45 density functional theory (DFT) methods have been used to predict structures and vibrational frequencies for Co4(CO)12, Rh4(CO)12, and Ir4(CO)12. The different DFT methods show a broad range of energy differences ΔE = ETd – EC3v. For Rh4(CO)12, none of the 45 DFT predictions is within 11 kcal/mol of the 2005 experimental value of 5.1 ± 0.6 kcal/mol reported by Allian and Garland ( Dalton Trans. 2005, 1957−1965). For the challenging Ir4(CO)12 molecule, 21 DFT methods predict the correct Td structure, while 24 DFT methods predict the C3v structure to lie lower in energy. This research reveals many peculiar problems in the computation of the vibrational frequencies for the all-terminal structure.
Co-reporter:Weihong Wu, Yunxiang Lu, Hairong Ding, Changjun Peng and Honglai Liu
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 2) pp:1339-1346
Publication Date(Web):12 Nov 2014
DOI:10.1039/C4CP04603J
Metal-containing ionic liquids (ILs) have been recognized as potential solvents, catalysts, catalyst precursors and reagents for many organic processes. In this work, several quantum-chemical parameters, including the surface electrostatic potential (Vs,max and Vs,min), the lowest surface average local ionization energy (s,min), and the electrostatic potential at the position of an atom (EPnuc), were adopted to understand the acidity/basicity of metal-containing ILs. Chlorometallate-based ILs show stronger acidity than conventional ILs, because of the increased electron-deficiency of the imidazole ring upon the incorporation of metal chloride. For the ILs with the Ag-coordinated cations, the acidity tends to attenuate while the basicity becomes stronger, as compared to traditional ILs. In addition, the regional Fukui function was also used to assess the molecular distribution of the Lewis acidity/basicity of the ILs under study. Overall, the introduction of metals into either the cations or the anions influences the acidity/basicity of ILs to a large degree, which would be beneficial for their certain applications, such as catalysis and extraction. We hope that the results presented here will assist in the development of novel metal-containing ILs with desirable properties.
Co-reporter:Shaoze Zhang, Zhaoqiang Chen, Yunxiang Lu, Zhijian Xu, Weihong Wu, Weiliang Zhu, Changjun Peng and Honglai Liu
RSC Advances 2015 vol. 5(Issue 91) pp:74284-74294
Publication Date(Web):26 Aug 2015
DOI:10.1039/C5RA13988K
Halogen bonds in imidazolium-based ion pairs have attracted recent research interest, due to their importance in the fields of anion recognition and ionic liquids. According to our survey of the Cambridge Structural Database (CSD), a number of crystal structures involving these specific halogen bonds were extracted. In this work, three different types of halogen bonding interactions, i.e. ion-pair halogen bonds, charge-assisted and neutral halogen bonds, in a series of dimeric complexes of imidazolium species were systematically studied at the M06-2x and B3LYP levels of theory. Ion-pair halogen bonds, despite being considerably stronger, show similar structural and energetic characteristics to common charge-assisted and neutral halogen bonds. To gain a deeper understanding of these interactions, the atoms in molecules (AIM), noncovalent interaction index (NCI), and energy decomposition analysis (EDA) calculations were undertaken. Most halogen bonds in imidazolium-based ion pairs have some covalent character, while the other two kinds of halogen bonds are weak electrostatic interactions. The attraction of ion-pair halogen bonds arises dominantly from electrostatic forces, while dispersion interaction plays a minor role. These two terms, however, contribute almost equally to the attraction of neutral halogen bonds. In addition to electrostatic attraction, induction interaction, which corresponds to charge transfer and mixing terms, also plays an important role in ion-pair and charge-assisted halogen bonds. The results presented should assist in the development of potent imidazolium-based anion receptors and novel halogenated ionic liquids with promising properties.
Pioneering synthetic research by the groups of Grutzmacher and Goicoechea have made possible the preparation of 2-phosphaethynolates (PCO–). The obvious question arises: can progress be made toward AsCO–, SbCO–, and BiCO–? Here the properties of all five anion congeners ECO– (E = N, P, As, Sb, Bi) were systematically investigated using ab initio coupled-cluster methods with correlation-consistent basis sets cc-pVXZ (X = D, T, Q). These anions exhibit linear structures with significant natural bond orbital negative charge on both the E and O atoms. These species should react with electrophiles via attack at either center. On going from nitrogen to bismuth, with the atomic radius increasing, the bond between E and C becomes weaker, while the C–O bond tends to be slightly stronger. By the time one gets to BiCO–, the C–O bond distance is 1.181 Å, indicating a very strong double bond. Relative to the PCO– anion, which is reactive toward several unsaturated compounds, the As/Sb/BiCO– anions may undergo cycloaddition more readily with unsaturated substrates. The dissociation energy of the E–C bond, except for that of NCO–, is predicted to be much less than that of the C–O bond. These dissociation energies are 76 kcal/mol (P––CO), 58 kcal/mol (As––CO), 37 kcal/mol (Sb––CO), and 28 kcal/mol (Bi––CO). Even the BiCO– anion should be achievable in the laboratory. The vibrational frequencies for these anions are predicted, and our results should assist in the experimental characterization and exploration of the heavier congeners ECO–.
Co-reporter:Qiaocui Shi, Hairong Su, Yingtao Liu, Weihong Wu, Yunxiang Lu
Computational and Theoretical Chemistry 2014 Volume 1027() pp:79-83
Publication Date(Web):1 January 2014
DOI:10.1016/j.comptc.2013.11.015
•Electron-donating substituents lead to stronger halogen bonds.•X⋯N interactions play a key role in XB-based crystal engineering.•Experimental evidence of the interactions was obtained from CSD.X⋯N interactions between halo-perfluorobenzenes and pyridine derivatives play a crucial role in halogen-bond-based crystal engineering and supramolecular architecture. In this work, a series of complexes of C6F5X (X = Br and I) with several pyridine molecules, which were extracted from crystal structures, have been calculated at the M05-2x level of theory. It was found that X⋯N interactions in the studied complexes are comparable in strength to medium strong hydrogen bonds. The introduction of electron-withdrawing substituents in pyridine attenuates X⋯N interactions, while the presence of electron-donating substituents enhances these halogen bonds. The quantum theory of atoms in molecules (QTAIM) and electrostatic potential (ESP) analyses were also employed to characterize these interactions. The results showed that electrostatic terms contribute significantly to the formation of X⋯N halogen bonds. Finally, some X-ray crystal structures retrieved from the Cambridge Structural Database (CSD) were selected to provide experimental evidence of present halogen bonds.Graphical abstract
Co-reporter:Weihong Wu, Yunxiang Lu, Yingtao Liu, Changjun Peng, Honglai Liu
Computational and Theoretical Chemistry 2014 Volume 1029() pp:21-25
Publication Date(Web):1 February 2014
DOI:10.1016/j.comptc.2013.12.008
•Substituents in electron donors have a slight effect on halogen bonding.•The presence of transition metals leads to much stronger halogen bonds.•Crystallographic evidence has been obtained from the CSD.According to our survey of the Cambridge Structural Database (CSD), a large number of X-ray crystal structures containing C–X⋯NCC/C–X⋯NCM halogen bonds were extracted. In this work, DFT/M06 calculations have been carried out to investigate the influence of substituents and transition metals on halogen bonding. It was found that electron-withdrawing/electron-donating substituents in electron donors slightly weaken/strengthen halogen bonds, while the introduction of transition metals leads to somewhat stronger halogen bonds. On the basis of electrostatic potential (ESP) analysis, these interactions are mainly electrostatic in nature, and moreover electrostatic forces play an important role in the enhancement or attenuation of halogen bonds with the presence of substituents and transition metals in electron donors. In addition, some X-ray crystal structures retrieved from the CSD were selected to provide experimental evidence of these halogen bonds.Graphical abstract
Co-reporter:Yanhua Wang, Jianying Tong, Weihong Wu, Yunxiang Lu
Computational and Theoretical Chemistry 2014 Volume 1049() pp:97-101
Publication Date(Web):1 December 2014
DOI:10.1016/j.comptc.2014.09.029
•Halogen bonds between I2 and ion pairs show some covalent character.•The interactions in ion pair systems belong to strong halogen bonds.•Ionic liquids involving halide anions have the potential for capture I2.To elucidate the efficient removal and reliable storage of radioactive iodine for ionic liquids, halogen bonds in the complexes of I2 with a series of ion pairs were investigated at the M06-2x level of theory in this work. Albeit halogen bonds in these complexes are somewhat weaker than those in the systems of bare anions, these interactions still belong to strong halogen bonds and show some covalent character. The strength of these interactions deceases in the following order: [Cl]− ≈ [Br]− ≈ [I]− > [Ac]− ≈ [NO3]− > [BF4]−, consistent with the experimentally determined trend of the removal efficiency for corresponding ionic liquids. In addition, the electrostatic potential, atoms in molecules, and charge transfer analyses were also undertaken for the complexes under study, to gain a deeper understanding of the nature of these interactions.
The Journal of Physical Chemistry A 2014 Volume 118(Issue 13) pp:2508-2518
Publication Date(Web):March 14, 2014
DOI:10.1021/jp4125167
Transition metal-containing ionic liquids (TM-ILs) have attracted a great deal of attention in recent years, due to their unique physical and chemical properties. In this work, several representative TM-ILs, such as the cations consisting of silver(I) center coodinated by two n-alkylimidazole ligands ([(Cnim)Ag(mim)]+) and the anions involving mercury(II) (HgCl3–), zinc(II) (ZnCl3–), and rhenium(VII) (ReO4–), were investigated using density functional theory calculations. First, the structural and energetic properties of the ion pairs for these TM-ILs have been examined in detail and compared with properties for conventional ILs. It was found that the interactions between the cations and anions, including hydrogen bonds and electrostatic interactions, in TM-ILs become weaker in strength than those in traditional ILs. In particular, the calculated geometric and energetic features compare fairly well with the experimental results, such as melting points and X-ray crystal structures of these TM-ILs. Then, the structures and energetics of ion-pair dimers for three ILs containing HgCl3–, ZnCl3–, and ReO4– were also explored, to gain a deeper understanding of the properties of TM-ILs. Finally, a survey of the Cambridge Structural Database (CSD) was undertaken to provide some crystallographic implications of TM-ILs.
Co-reporter:Haiying Li, Yunxiang Lu, Weihong Wu, Yingtao Liu, Changjun Peng, Honglai Liu and Weiliang Zhu
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 12) pp:4405-4414
Publication Date(Web):28 Jan 2013
DOI:10.1039/C3CP44649B
In recent years, several specific imidazolium-based ionic liquids with halogen substituents on the imidazole ring as well as on the alkyl chains have been reported. In this work, noncovalent interactions in four halogenated ionic liquids, i.e. 2-bromo-/iodo- and 4,5-dibromo-/diiodo-1,3-dimethylimidazolium trifluoromethanesulfonates, were systematically investigated using density functional theory calculations. The structural and energetic properties of the ion pairs for such ionic liquids have been fully examined and compared with the non-halogenated ones. It was found that C–X⋯O halogen bonds, C–H⋯O hydrogen bonds, and electrostatic interactions with the anion located over the imidazole ring in the ion pairs. In addition, the structures and energetics of two ion pairs for such ionic liquids were also explored to reproduce experimental observations. The halogen-bonded ring structures and the conformers with the concurrent C–H⋯O and C–X⋯O contacts were predicted, consistent with the X-ray crystal structures of corresponding organic salts. Finally, the implications of the observed structural and energetic features of ion pairs on the design of halogen-bonding ionic liquids were discussed. The results presented herein should provide useful information in the development of novel halogenated ionic liquids used for specific tasks ranging from organic synthesis to gas absorption.
Co-reporter:Yanhua Wang, Weihong Wu, Yingtao Liu, Yunxiang Lu
Chemical Physics Letters 2013 Volume 578() pp:38-42
Publication Date(Web):18 July 2013
DOI:10.1016/j.cplett.2013.06.005
Highlights•The presence of coordination forces leads to much stronger halogen bonds.•Experimental evidence has been obtained from the CSD.•Electrostatic terms play an important role in the enhancement of halogen bonds.Density functional theory calculations at the level of M06 have been carried out to investigate the influence of transition metal coordinate on halogen bonding. It was found that the introduction of coordination forces leads to much stronger halogen bonds. This effect has been analyzed in detail by the geometric, energetic, electrostatic potential, and AIM properties of the complexes. In addition, some crystal structures extracted from the Cambridge Structural Database were selected to provide experimental evidence of the combination of the two interactions.Graphical abstract
Chemical Physics Letters 2013 Volume 582() pp:49-55
Publication Date(Web):4 September 2013
DOI:10.1016/j.cplett.2013.07.048
•Weak cooperative or diminutive effects occur in the studied complexes.•The different energetic effects are explained by the direction of charge transfer.•Experimental evidence has been obtained from the CSD.MP2 and M05-2x calculations have been carried out to investigate the interplay between X–π and X–N halogen bonds. Weak cooperative or diminutive effects are observed in the complexes where the two interactions coexist. For comparison, the mutual influence between H–π interactions and halogen bonds was also examined and the same energetic effects take place. These effects have been analyzed in detail by the structural, energetic, and AIM properties of the complexes. In addition, some crystal structures extracted from the Cambridge Structural Database were selected to provide experimental evidence of the combination of the two interactions.
Co-reporter:Yanhua Wang, Haiying Li, Yingtao Liu, Weihong Wu, Yunxiang Lu
Computational and Theoretical Chemistry 2013 Volume 1026() pp:1-6
Publication Date(Web):15 December 2013
DOI:10.1016/j.comptc.2013.10.012
•Weak energetic effects are observed in the studied crystal structures.•Calculated energetic results agree well with the geometric features.•Energetic effects are much more complicated in crystal structures.The interplay between halogen and hydrogen bonds in certain crystal structures of halo-perfluorobenzenes (X-PFCs) and pyrazine molecules has been investigated by means of the DFT/M05-2x method. Very weak energetic effects are observed in the studied complexes, which indicates that the two kinds of noncovalent interactions have some additive aspects. These effects have been fully examined by the geometric and energetic features of the complexes.Graphical abstract
The Journal of Physical Chemistry B 2013 Volume 117(Issue 17) pp:4827-4835
Publication Date(Web):April 1, 2013
DOI:10.1021/jp4001658
Database survey in this study revealed that about one-third of the protein structures deposited in the Protein Data Bank (PDB) contain arginine–arginine (Arg–Arg) pairing with a carbon···carbon (CZ···CZ) interaction distance less than 5 Å. All the Arg–Arg pairings were found to bury in a polar environment composed of acidic residues, water molecules, and strong polarizable or negatively charged moieties from binding site or bound ligand. Most of the Arg–Arg pairings are solvent exposed and 68.3% Arg–Arg pairings are stabilized by acidic residues, forming Arg–Arg–Asp/Glu clusters. Density functional theory (DFT) was then employed to study the effect of environment on the pairing structures. It was revealed that Arg–Arg pairings become thermodynamically stable (about −1 kcal/mol) as the dielectric constant increases to 46.8 (DMSO), in good agreement with the results of the PDB survey. DFT calculations also demonstrated that perpendicular Arg–Arg pairing structures are favorable in low dielectric constant environment, while in high dielectric constant environment parallel structures are favorable. Additionally, the acidic residues can stabilize the Arg–Arg pairing structures to a large degree. Energy decomposition analysis of Arg–Arg pairings and Arg–Arg–Asp/Glu clusters showed that both solvation and electrostatic energies contribute significantly to their stability. The results reported herein should be very helpful for understanding Arg–Arg pairing and its application in drug design.
Co-reporter:Haiying Li, Yunxiang Lu, Yingtao Liu, Xiang Zhu, Honglai Liu and Weiliang Zhu
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 28) pp:9948-9955
Publication Date(Web):24 May 2012
DOI:10.1039/C2CP41149K
According to our survey of the Cambridge Structural Database (CSD), a great number of crystal structures, in which halogen bonds and aromatic stacking interactions are present and play an important role in crystal packing, have been extracted. In this work, ab initio calculations at the MP2 level of theory were performed to investigate the mutual influence between halogen bonds and π–π stacking interactions. Different energetic effects are observed in the studied complexes where the two kinds of noncovalent interactions coexist, which can be rationalized by the direction of charge transfer for the two interactions. These effects have been analyzed in detail in terms of the structural, energetic, and charge transfer properties of the complexes. In addition, the quantum theory of atoms in molecules (QTAIM) was also employed to characterize the interactions and to examine the strengthening or weakening of the interactions, depending on the variations of electron density on the bond and cage critical points. Finally, certain crystal structures retrieved from the CSD have been selected to provide experimental evidence of the combination of the two interactions.
The interplay between halogen bonds and cation–π interactions is investigated by ab initio calculations at the MP2 level of theory. Different energetic effects are observed in the studied complexes in which halogen bonds and cation–π interactions coexist, which can be ascribed to the direction of charge transfer for the two interactions. These effects are analyzed in detail in terms of the structural, energetic, and charge-transfer properties of the complexes. In addition, the quantum theory of atoms in molecules is employed to characterize the interactions and to examine their enhancement and attenuation in terms of the variations in electron density at the bond and cage critical points. Finally, experimental evidence for a combination of the two interactions is obtained from the Cambridge Structural Database.
The Journal of Physical Chemistry A 2012 Volume 116(Issue 10) pp:2591-2597
Publication Date(Web):February 21, 2012
DOI:10.1021/jp212522k
Energetic effects between halogen bonds and anion-π or lone pair-π interactions have been investigated by means of ab initio MP2 calculations. 1,4-diiodo-perfluorobenzene, a very effective building block for crystal engineering based on halogen bonding, is selected in this work both as electron-deficient π aromatic ring and as halogen bond donor. Additive and diminutive effects are observed when halogen bonds and anion-π/lone pair-π interactions coexist in the same complex, which can be ascribed to the same direction of charge transfer for the two interactions. These effects have been analyzed in detail by the structural, energetic, and AIM properties of the complexes. Finally, experimental evidence of the combination of the interactions has been obtained from the Cambridge Structural Database.
Co-reporter:HaiYing Li;Xiang Zhu;ChangJun Peng;Jun Hu
Science China Chemistry 2012 Volume 55( Issue 8) pp:1566-1572
Publication Date(Web):2012 August
DOI:10.1007/s11426-012-4648-0
Halogen bonding interactions between several halogenated ion pairs and CO2 molecules have been investigated by means of density functional theory calculations. To account for the influence of solvent environment, the implicit polarized continuum model was also employed. The bromide and iodide cations of ionic liquids (ILs) under study can interact with CO2 molecules via X…O interactions, which become much stronger in strength than those in the complexes of iodo-perfluorobenzenes, very effective halogen bond donors, with CO2 molecules. Such interactions, albeit somewhat weaker in strength, are also observed between halogenated ion pairs and CO2 molecules. Thus, the solubility of CO2 may be improved when using halogenated ILs, as a result of the formation of X…O halogen bonds. Under solvent effects, the strength of the interactions tends to be weakened to some degree, with a concomitant elongation of intermolecular distances. The results presented here would be very useful in the design and synthesis of novel and potent ILs for CO2 physical absorption.
Co-reporter:Yunxiang Lu, Haiying Li, Xiang Zhu, Weiliang Zhu, and Honglai Liu
The Journal of Physical Chemistry A 2011 Volume 115(Issue 17) pp:4467-4475
Publication Date(Web):April 5, 2011
DOI:10.1021/jp111616x
A systematic study of halogen bonding interactions in gas phase and in solution was carried out by means of quantum chemical DFT/B3LYP method. Three solvents with different polarities (chloroform, acetone, and water) were selected, and solvation effects were considered using the polarized continuum model (PCM). For charged halogen-bonded complexes, the strength of the interactions tends to significantly weaken in solution, with a concomitant elongation of intermolecular distances. For neutral systems, halogen bond distances are shown to shorten and the interaction energies change slightly. Computations also reveal that in the gas phase the binding affinities decrease in the order Cl− > Br− > I−, while in solution the energy gaps of binding appear limited for the three halide anions. According to free energy results, many systems under investigation are stable in solution. Particularly, calculated free energies of formation of the complexes correlate well with halogen-bonding association constants determined experimentally. The differences of the effects of solvent upon halogen and hydrogen bonding were also elucidated. This study can establish fundamental characteristics of halogen bonding in media, which would be very helpful for applying this noncovalent interaction in medicinal chemistry and material design.
Weak S···O bonding, a specific noncovalent interaction, plays crucial roles in fields as diverse as molecular recognition, crystal engineering, and biological systems. This article presents an ab initio investigation of a series of dimeric complexes formed between formaldehyde and several sulfur-containing molecules as electron accepters. The bond-length change, interaction energy, topological property of the electron density, and charge transfer of these S···O bonds have been systematically investigated. Moreover, a comprehensive search for nonbonded S···O interactions in proteins was also performed. It was found that the O atom shows a strong intrinsic tendency to approach S from the backside of the R–S bond (in the σs* direction); the S atom tends to approach the O atom either from the orientation of the lone pair of O (in the no direction) or from the vertical direction (in the πo direction). Besides, the linearity of this interaction was further substantiated by the statistical study. As suggested by the results presented in this study, S···O contacts may control protein structures to some extent and the unique directional properties of S···O interactions could be applied in supermolecular assembly and biological design.
Co-reporter:Haiying Li, Yunxiang Lu, Yingtao Liu, Xiang Zhu, Honglai Liu and Weiliang Zhu
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 28) pp:NaN9955-9955
Publication Date(Web):2012/05/24
DOI:10.1039/C2CP41149K
According to our survey of the Cambridge Structural Database (CSD), a great number of crystal structures, in which halogen bonds and aromatic stacking interactions are present and play an important role in crystal packing, have been extracted. In this work, ab initio calculations at the MP2 level of theory were performed to investigate the mutual influence between halogen bonds and π–π stacking interactions. Different energetic effects are observed in the studied complexes where the two kinds of noncovalent interactions coexist, which can be rationalized by the direction of charge transfer for the two interactions. These effects have been analyzed in detail in terms of the structural, energetic, and charge transfer properties of the complexes. In addition, the quantum theory of atoms in molecules (QTAIM) was also employed to characterize the interactions and to examine the strengthening or weakening of the interactions, depending on the variations of electron density on the bond and cage critical points. Finally, certain crystal structures retrieved from the CSD have been selected to provide experimental evidence of the combination of the two interactions.
Co-reporter:Weihong Wu, Yunxiang Lu, Hairong Ding, Changjun Peng and Honglai Liu
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 2) pp:NaN1346-1346
Publication Date(Web):2014/11/12
DOI:10.1039/C4CP04603J
Metal-containing ionic liquids (ILs) have been recognized as potential solvents, catalysts, catalyst precursors and reagents for many organic processes. In this work, several quantum-chemical parameters, including the surface electrostatic potential (Vs,max and Vs,min), the lowest surface average local ionization energy (s,min), and the electrostatic potential at the position of an atom (EPnuc), were adopted to understand the acidity/basicity of metal-containing ILs. Chlorometallate-based ILs show stronger acidity than conventional ILs, because of the increased electron-deficiency of the imidazole ring upon the incorporation of metal chloride. For the ILs with the Ag-coordinated cations, the acidity tends to attenuate while the basicity becomes stronger, as compared to traditional ILs. In addition, the regional Fukui function was also used to assess the molecular distribution of the Lewis acidity/basicity of the ILs under study. Overall, the introduction of metals into either the cations or the anions influences the acidity/basicity of ILs to a large degree, which would be beneficial for their certain applications, such as catalysis and extraction. We hope that the results presented here will assist in the development of novel metal-containing ILs with desirable properties.
Co-reporter:Haiying Li, Yunxiang Lu, Weihong Wu, Yingtao Liu, Changjun Peng, Honglai Liu and Weiliang Zhu
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 12) pp:NaN4414-4414
Publication Date(Web):2013/01/28
DOI:10.1039/C3CP44649B
In recent years, several specific imidazolium-based ionic liquids with halogen substituents on the imidazole ring as well as on the alkyl chains have been reported. In this work, noncovalent interactions in four halogenated ionic liquids, i.e. 2-bromo-/iodo- and 4,5-dibromo-/diiodo-1,3-dimethylimidazolium trifluoromethanesulfonates, were systematically investigated using density functional theory calculations. The structural and energetic properties of the ion pairs for such ionic liquids have been fully examined and compared with the non-halogenated ones. It was found that C–X⋯O halogen bonds, C–H⋯O hydrogen bonds, and electrostatic interactions with the anion located over the imidazole ring in the ion pairs. In addition, the structures and energetics of two ion pairs for such ionic liquids were also explored to reproduce experimental observations. The halogen-bonded ring structures and the conformers with the concurrent C–H⋯O and C–X⋯O contacts were predicted, consistent with the X-ray crystal structures of corresponding organic salts. Finally, the implications of the observed structural and energetic features of ion pairs on the design of halogen-bonding ionic liquids were discussed. The results presented herein should provide useful information in the development of novel halogenated ionic liquids used for specific tasks ranging from organic synthesis to gas absorption.






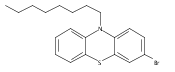
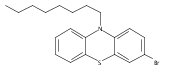
![PYRROLO[3,4-C]PYRROLE-1,4-DIONE, 3,6-BIS(3-FLUOROPHENYL)-2,5-DIHYDRO-](http://img.cochemist.com/ccimg/850700/850698-62-3.png)
![PYRROLO[3,4-C]PYRROLE-1,4-DIONE, 3,6-BIS(3-FLUOROPHENYL)-2,5-DIHYDRO-](http://img.cochemist.com/ccimg/850700/850698-62-3_b.png)
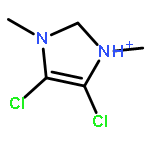

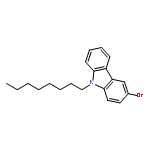




![DIAZENE, BIS[4-(BROMOMETHYL)PHENYL]-](http://img.cochemist.com/ccimg/97700/97654-99-4.png)
![DIAZENE, BIS[4-(BROMOMETHYL)PHENYL]-](http://img.cochemist.com/ccimg/97700/97654-99-4_b.png)
![Pyrrolo[3,4-c]pyrrole-1,4-dione, 2,5-dibutyl-2,5-dihydro-3,6-diphenyl-](/data/chemimg/3612600/96159-01-2.png)
![Pyrrolo[3,4-c]pyrrole-1,4-dione, 2,5-dibutyl-2,5-dihydro-3,6-diphenyl-](/data/chemimg/3612600/96159-01-2_b.png)
![Pyrrolo[3,4-c]pyrrole-1,4-dione,2,5-dihydro-3,6-bis(4-methylphenyl)-](/data/chemimg/976000/84632-66-6.png)
![Pyrrolo[3,4-c]pyrrole-1,4-dione,2,5-dihydro-3,6-bis(4-methylphenyl)-](/data/chemimg/976000/84632-66-6_b.png)
![Pyrrolo[3,4-c]pyrrole-1,4-dione, 3,6-bis(4-fluorophenyl)-2,5-dihydro-](/data/chemimg/3448900/84632-57-5.png)
![Pyrrolo[3,4-c]pyrrole-1,4-dione, 3,6-bis(4-fluorophenyl)-2,5-dihydro-](/data/chemimg/3448900/84632-57-5_b.png)