Co-reporter:Pei Cai, Si-Qiang Fang, Xue-Lian Yang, Jia-Jia Wu, Qiao-Hong Liu, Hao Hong, Xiao-Bing Wang, and Ling-Yi Kong
ACS Chemical Neuroscience November 15, 2017 Volume 8(Issue 11) pp:2496-2496
Publication Date(Web):August 14, 2017
DOI:10.1021/acschemneuro.7b00257
A novel series of donepezil-trolox hybrids were designed, synthesized, and evaluated as multifunctional ligands against Alzheimer’s disease (AD). Biological assays showed that these derivatives possessed moderate to good inhibitory activities against acetylcholinesterase (AChE) and monoamine oxidase B (MAO-B) as well as remarkable antioxidant effects. The optimal compound 6d exhibited balanced functions with good inhibition against hAChE (IC50 = 0.54 μM) and hMAO-B (IC50 = 4.3 μM), significant antioxidant activity (41.33 μM IC50 by DPPH method, 1.72 and 1.79 trolox equivalent by ABTS and ORAC methods), excellent copper chelation, and Aβ1–42 aggregation inhibition effect. Furthermore, cellular tests indicated that 6d has very low toxicity and is capable of combating oxidative toxin (H2O2, rotenone, and oligomycin-A) induced neurotoxicity. Most importantly, oral administration of 6d demonstrated notable improvements on cognition and spatial memory against scopolamine-induced acute memory deficit as well as d-galactose (d-gal) and AlCl3 induced chronic oxidative stress in a mouse model without acute toxicity and hepatotoxicity. In summary, both in vitro and in vivo results suggested that 6d is a valuable candidate for the development of a safe and effective anti-Alzheimer’s drug.Keywords: acetylcholinesterase inhibitors; Alzheimer’s disease; antioxidant; cognitive improvement; neuroprotection; β-amyloid aggregation;
Co-reporter:Jia-Jia Wu, Ting Ma, Zhi-Min Wang, Wen-Jun Xu, Xue-Lian Yang, Jian-Guang Luo, Ling-Yi Kong, Xiao-Bing Wang
Journal of Functional Foods 2017 Volume 28() pp:205-214
Publication Date(Web):January 2017
DOI:10.1016/j.jff.2016.11.022
•Six xanthone analogues (1–6) were obtained in an environmental friendly way.•Compound 1 with a peroxide group exhibited potent cytotoxic activity.•Compound 1 induced G2/M arrest and activated caspase-dependent apoptotic pathway.•Compound 1 inhibited the ROS-mediated and MAPK signalling pathway.A new peroxide of xanthone (1), together with five analogues (2–6), was obtained from horseradish peroxidase (HRP) catalysed biotransformation of α-mangostin. In order to increase the yield of compound 1, the environmental pH was succinctly adjusted, and as a result, compound 1 was formed with considerable selectivity. The anticancer potential of compound 1, a pentacyclic xanthone with a 1,2-dioxolane ring, was investigated due to its potent cytotoxic activity. The results showed that apoptosis induced by compound 1 in human hepatocellular carcinoma (HepG2) cell was associated with activation of caspase-dependent apoptotic pathway, the generation of reactive oxygen species (ROS) and the activation of c-Jun N-terminal kinases (JNK). In summary, compound 1 with a peroxide group was biosynthesized in considerable yield through pH-switched biotransformation catalysed by HRP, which could be further developed as a promising candidate in the treatment of liver cancer.
Co-reporter:Qiao-Hong Liu;Jia-Jia Wu;Fan Li;Pei Cai;Xue-Lian Yang;Ling-Yi Kong
MedChemComm (2010-Present) 2017 vol. 8(Issue 7) pp:1459-1467
Publication Date(Web):2017/07/19
DOI:10.1039/C7MD00199A
A series of homoisoflavonoid derivatives was designed, synthesized and evaluated as potential multi-functional anti-Alzheimer's agents for their inhibitory activity on cholinesterase and monoamine oxidase. Among them, compound 16 showed moderate acetylcholinesterase (AChE) inhibitory activity (eeAChE IC50 = 0.89 ± 0.02 μM; hAChE IC50 = 0.657 ± 0.002 μM) and significant monoamine oxidase B (MAO-B) inhibitory activity (hMAO-B IC50 = 0.0372 ± 0.0002 μM). Kinetic analysis of AChE, MAO-B inhibition and molecular modeling studies revealed that compound 16 is a dual binding site inhibitor of AChE and noncompetitive inhibitor of MAO-B. Furthermore, 16 could penetrate through the blood–brain barrier (BBB) in vitro. Most importantly, oral administration of 16 demonstrated no marked signs of acute toxicity and it could significantly reverse scopolamine-induced memory impairment in mice. These results suggested that compound 16 is a promising multifunctional drug candidate with potential effect for the treatment of Alzheimer's disease.
Co-reporter:Fan Li, Jia-Jia Wu, Jin Wang, Xue-Lian Yang, Pei Cai, Qiao-Hong Liu, Ling-Yi Kong, Xiao-Bing Wang
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 14(Issue 14) pp:
Publication Date(Web):15 July 2017
DOI:10.1016/j.bmc.2017.05.027
•A series of chromone derivatives were designed, synthesized and evaluated.•Most of the target compounds have both MAOs inhibition activities, antioxidant activity and biometal chelating ability.•Compound s19 exhibits good inhibitory potency for inhibition of MAOs (IC50 value of 5.12 μM for hMAO-A and 0.816 μM for hMAO-B).•Compound s19 could across the blood–brain barrier.In a continuing effort to develop multitargeted compounds as potential treatment agents against Alzheimer’s disease (AD), a series of chromone derivatives were designed, synthesized and evaluated. In vitro assay indicated that most of the target compounds have both MAOs inhibition activities, antioxidant activity and biometal chelating ability. Especially, compound s19 exhibits good inhibitory potency for inhibition of MAOs (IC50 value of 5.12 μM for hMAO-A and 0.816 μM for hMAO-B), moderate inhibition of Aβ aggregation (75.1% at 20 μM), metal chelation, control of ROS generation and antioxidant activity (ORAC = 3.62). In addition, s19 could reduce PC12 cells death induced by oxidative stress and penetrate the blood–brain barrier (BBB). Taken together, these results suggested that s19 might be a promising multitargeted compound for AD treatment.Download high-res image (123KB)Download full-size image
Co-reporter:Hua-Li Yang, Pei Cai, Qiao-Hong Liu, Xue-Lian Yang, Si-Qiang Fang, Yan-Wei Tang, Cheng Wang, Xiao-Bing Wang, Ling-Yi Kong
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 21(Issue 21) pp:
Publication Date(Web):1 November 2017
DOI:10.1016/j.bmc.2017.08.048
•Novel multi-target-directed ligands for Alzheimer's disease were designed and synthesized.•Most of the target compounds have inhibitory activities against amyloid-β (Aβ) aggregation and hMAO-B as well as remarkable antioxidant effects.•Compound 5 exhibited excellent potency for inhibition of self-induced Aβ1–42 aggregation (91.3 ± 2.1%, 25 μM) and inhibition of hMAO-B (IC50, 1.73 ± 0.39 μM).•Compound 5 showed antioxidant activitities, metal-chelating properties, neuroprotective effects, significant in vitro anti-inflammatory activity and potent BBB penetration.A series of salicyladimine derivatives were designed, synthesized and evaluated as multi-target-directed ligands for the treatment of Alzheimer’s disease (AD). Biological activity results demonstrated that some derivatives possessed significant inhibitory activities against amyloid-β (Aβ) aggregation and human monoamine oxidase B (hMAO-B) as well as remarkable antioxidant effects and low cell toxicity. The optimal compound, 5, exhibited excellent potency for inhibition of self-induced Aβ1–42 aggregation (91.3 ± 2.1%, 25 μM), inhibition of hMAO-B (IC50, 1.73 ± 0.39 μM), antioxidant effects (43.4 ± 2.6 μM of IC50 by DPPH method, 0.67 ± 0.06 trolox equivalent by ABTS method), metal chelation and BBB penetration. Furthermore, compound 5 had neuroprotective effects against ROS generation, H2O2-induced apoptosis, 6-OHDA-induced cell injury, and a significant in vitro anti-inflammatory activity. Collectively, these findings highlighted that compound 5 was a potential balanced multifunctional neuroprotective agent for the development of anti-AD drugs.Download high-res image (88KB)Download full-size image
Co-reporter:Jin Wang, Pei Cai, Xue-Lian Yang, Fan Li, Jia-Jia Wu, Ling-Yi Kong, Xiao-Bing Wang
European Journal of Medicinal Chemistry 2017 Volume 139(Volume 139) pp:
Publication Date(Web):20 October 2017
DOI:10.1016/j.ejmech.2017.07.077
•Cinnamamide-dibenzylamine hybrids were designed, synthesized and evaluated.•The new hybrids were obtained by fusing cinnamamide with AP2238.•Most of the hybrids exhibited a significant ability to inhibit ChEs.•Some had the ability of anti-Aβ aggregation, antioxidant and biometal chelating.•7f is a balance multitargeted candidate worthy of further investigation.By using fragments endowed with interesting and complementary properties for the treatment of Alzheimer's disease (AD), a novel series of cinnamamide-dibenzylamine hybrids have been designed, synthesized, and evaluated biologically. In vitro assay indicated that most of the target compounds exhibited a significant ability to inhibit ChEs, strong potency inhibitory of self-induced β-amyloid (Aβ) aggregation and to act as potential antioxidants and biometal chelators. A Lineweaver-Burk plot and molecular modeling study showed that compound 7f targeted both the CAS and PAS of AChE. In addition, compound 7f could chelate metal ions, reduce PC12 cells death induced by oxidative stress and penetrate the blood–brain barrier (BBB). Overall, all of these outstanding in vitro results in combination with promising in vivo outcomes highlighted derivative 7f as the lead structure worthy of further investigation.Download high-res image (130KB)Download full-size image
Co-reporter:Zhi-Min Wang, Pei Cai, Qiao-Hong Liu, Ding-Qiao Xu, Xue-Lian Yang, Jia-Jia Wu, Ling-Yi Kong, Xiao-Bing Wang
European Journal of Medicinal Chemistry 2016 Volume 123() pp:282-297
Publication Date(Web):10 November 2016
DOI:10.1016/j.ejmech.2016.07.052
•A series of donepezil derivatives were designed and synthesized.•Most of the compounds showed potent and selective AChE inhibitory activities.•Some compounds acted as multifunctional agents with AChE inhibition, metal chelation, antioxidation and Aβ interaction.•Compounds 3d and 5c showed significantly reversed scopolamine-induced memory deficit in mice.A series of novel donepezil derivatives was designed, synthesized and evaluated as multifunctional acetylcholinesterase (AChE) inhibitors for the treatment of Alzheimer's disease (AD). The screening results indicated that most of the compounds exhibited potent inhibition of AChE with IC50 values in the nanomolar range. Moreover, these derivatives displayed good antioxidant, Aβ interaction, blood-brain barrier penetration (PAMPA-BBB+) and ADMET properties (in silico). Among them, 5c demonstrated excellent AChE inhibition (IC50: 85 nM for eeAChE, 73 nM for hAChE), metal chelation, and inhibitory effects on self-induced, hAChE-induced and Cu2+-induced Aβ1-42 aggregation (18.5%, 72.4% and 46.3%, at 20 μM). Kinetic analysis and molecular modeling studies suggested that 5c could bind simultaneously to the catalytic active site (CAS) and peripheral anionic site (PAS) of AChE. More importantly, 5c exhibited significant neuroprotective potency against Aβ1-42-induced PC12 cell injury. Furthermore, the step-through passive avoidance test showed 5c significantly reversed scopolamine-induced memory deficit and no hepatotoxicity in mice. These results indicated that 5c might be a promising drug candidate for AD therapy.
Co-reporter:Jin Wang, Zhi-Min Wang, Xue-Mei Li, Fan Li, Jia-Jia Wu, Ling-Yi Kong, Xiao-Bing Wang
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 18) pp:4324-4338
Publication Date(Web):15 September 2016
DOI:10.1016/j.bmc.2016.07.025
A novel series of compounds obtained by fusing the acetylcholinesterase (AChE) inhibitor donepezil and the antioxidant melatonin were designed as multi-target-directed ligands for the treatment of Alzheimer’s disease (AD). In vitro assay indicated that most of the target compounds exhibited a significant ability to inhibit acetylcholinesterase (eeAChE and hAChE), butyrylcholinesterase (eqBuChE and hBuChE), and β-amyloid (Aβ) aggregation, and to act as potential antioxidants and biometal chelators. Especially, 4u displayed a good inhibition of AChE (IC50 value of 193 nM for eeAChE and 273 nM for hAChE), strong inhibition of BuChE (IC50 value of 73 nM for eqBuChE and 56 nM for hBuChE), moderate inhibition of Aβ aggregation (56.3% at 20 μM) and good antioxidant activity (3.28 trolox equivalent by ORAC assay). Molecular modeling studies in combination with kinetic analysis revealed that 4u was a mixed-type inhibitor, binding simultaneously to catalytic anionic site (CAS) and the peripheral anionic site (PAS) of AChE. In addition, 4u could chelate metal ions, reduce PC12 cells death induced by oxidative stress and penetrate the blood–brain barrier (BBB). Taken together, these results strongly indicated the hybridization approach is an efficient strategy to identify novel scaffolds with desired bioactivities, and further optimization of 4u may be helpful to develop more potent lead compound for AD treatment.
Co-reporter:Zhimin Wang, Jiajia Wu, Xuelian Yang, Pei Cai, Qiaohong Liu, Kelvin D.G. Wang, Lingyi Kong, Xiaobing Wang
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 22) pp:5929-5940
Publication Date(Web):15 November 2016
DOI:10.1016/j.bmc.2016.09.050
•A series of representative benzyloxy substituted MAO-B inhibitors were synthesized and evaluated.•Compounds 5–8, 12–17, 25, 28 and 31 exhibited neuroprotective effects against 6-OHDA- and rotenone-treated PC12 cells.•Compound 13 showed neuroprotective effects against neurotoxins-induced ROS accumulation and apoptosis.•Compound 13 might be promising candidates for the therapy of Parkinson’s disease.The benzyloxy substituted small molecules are well-known highly potent monoamine oxidase B inhibitors, but their therapeutic potential against Parkinson’s disease have not been investigated in detail. In this paper, a series of representative benzyloxy substituted derivatives were synthesized and evaluated for MAO-A/B inhibition. In addition, their neuroprotective effects were investigated in 6-OHDA- and rotenone-treated PC12 cells. It was observed that most of the compounds exhibited a marked increase in survival of PC12 cells which treated with the neurotoxins. Among them, 13 exhibited remarkable and balanced neuroprotective potency. The protective effects of 13 against neurotoxins-induced apoptosis were confirmed with flow cytometry and staining methods. Furthermore, 13 also showed good BBB permeability and low toxicity according to in vitro BBB prediction and in vivo acute toxicity test. The results indicated that 13 is an effective and promising candidate to be further developed as disease-modifying drug for Parkinson’s disease therapy.
Co-reporter:Zhi-Min Wang, Sai-Sai Xie, Xue-Mei Li, Jia-Jia Wu, Xiao-Bing Wang and Ling-Yi Kong
RSC Advances 2015 vol. 5(Issue 86) pp:70395-70409
Publication Date(Web):18 Aug 2015
DOI:10.1039/C5RA13594J
A series of 3-Schiff base-4-hydroxycoumarin derivatives (1–20) have been designed, synthesized and evaluated as multifunctional agents against Alzheimer's disease. In vitro studies indicated that most of the derivatives exhibited significant abilities to inhibit monoamine oxidase (MAO), self-induced and Cu2+-induced β-amyloid (Aβ1–42) aggregati006Fn, as well acting as potential biometal chelators and antioxidants. Moreover, these derivatives were capable of decreasing reactive oxygen species (ROS) and showed good neuroprotective effects in PC12 cells and could penetrate the central nervous system (CNS). In particular, compound 4 exhibited high potency to monoamine oxidase (IC50, 0.673 μM for hMAO-A, 0.711 μM for hMAO-B), good antioxidant activity (1.34 trolox equivalents by ABTS method, 45.8 μM of IC50 by DPPH method), and it also displayed a significant ability to inhibit self-induced and Cu2+-induced Aβ1–42 aggregation (60.1%, 20 μM and 45.7%, 50 μM). Taken together, these results suggested that compound 4 might be a promising lead compound with balanced properties for AD treatment.
Co-reporter:Zhi-Min Wang, Xue-Mei Li, Gui-Min Xue, Wei Xu, Xiao-Bing Wang and Ling-Yi Kong
RSC Advances 2015 vol. 5(Issue 126) pp:104122-104137
Publication Date(Web):10 Dec 2015
DOI:10.1039/C5RA22296F
Considering the complex etiology of Alzheimer’s disease (AD), multifunctional agents may exhibit important properties against this disease. A series of 6-substituted 3-arylcoumarins (5a–t) were designed, synthesized and evaluated as cholinesterase (ChE) and monoamino oxidase (MAO) inhibitors. Among them, compounds 5o [IC50, 195 nM for human acetylcholinesterase (hAChE), selectivity index, SI (human butyrylcholinesterase, hBuChE/hAChE) = 145; IC50, 63.5 nM for human monoamine oxidase-B (hMAO-B), SI (human monoamine oxidase-A, hMAO-A/hMAO-B) = 25] and 5p [IC50, 185 nM for hAChE, SI (hBuChE/hAChE) = 182; IC50, 196 nM for hMAO-B, SI (hMAO-A/hMAO-B) > 510] were found to selectively inhibit both hAChE and hMAO-B with IC50 values in the nanomolar range. The abilities of 5o and 5p to bind to hAChE and hMAO-B were confirmed by molecular docking and kinetic studies. Moreover, 5o and 5p were found to exhibit significant inhibition of self-induced Aβ42 aggregation (61% and 52%, at 20 μM), have antioxidant properties (0.81 and 1.17 trolox equivalent by ABTS assay), provide neuroprotection against Aβ42-induced cytotoxicity and blood–brain barrier (BBB) penetration capacity (PAMPA-BBB+), and are thus potential anti-Alzheimer agents with balanced activities. Overall, the study provided meaningful information for further development of multifunctional drugs for AD therapy.
Co-reporter:Zhi-Min Wang, Xue-Mei Li, Wei Xu, Fan Li, Jin Wang, Ling-Yi Kong and Xiao-Bing Wang
MedChemComm 2015 vol. 6(Issue 12) pp:2146-2157
Publication Date(Web):21 Oct 2015
DOI:10.1039/C5MD00357A
Two series of acetophenone derivatives have been designed, synthesized and evaluated for human monoamine oxidase A and B inhibitory activity in vitro. Most of the tested compounds acted preferentially on MAO-B with IC50 values in the nanomolar range and weak or no inhibition of MAO-A. In particular, compounds 1j (IC50 = 12.9 nM) and 2e (IC50 = 11.7 nM) were the most potent MAO-B inhibitors being 2.76- and 2.99-fold more active than selegiline. In addition, the structure–activity relationships for MAO-B inhibition indicated that substituents at C3 and C4 of the acetophenone moiety, particularly with the halogen substituted benzyloxy, were more favorable for MAO-B inhibition. Molecular docking and kinetic studies have been carried out to explain the binding modes of MAO-B with the acetophenone derivatives. Furthermore, the representative compounds 1j and 2e showed low neurotoxicity in SH-SY5Y cells. It may be concluded that the acetophenone derivatives could be used to develop promising lead compounds for treating neurodegenerative diseases.
Co-reporter:Ya-Jian Hu, Xiao-Bing Wang, Su-Yi Li, Sai-Sai Xie, Kelvin D.G. Wang, Ling-Yi Kong
Tetrahedron Letters 2015 Volume 56(Issue 1) pp:105-108
Publication Date(Web):1 January 2015
DOI:10.1016/j.tetlet.2014.11.026
Co-reporter:Jin-Shuai Lan, Long-Fei Pan, Sai-Sai Xie, Xiao-Bing Wang and Ling-Yi Kong
MedChemComm 2015 vol. 6(Issue 4) pp:592-600
Publication Date(Web):15 Dec 2014
DOI:10.1039/C4MD00437J
A new series of 6-methyl-3-phenylcoumarins (3a–c and 5a–o) and 6-methyl-3-heteroarylcoumarins (5p–s) have been designed, synthesized and evaluated as monoamine oxidase inhibitors. The results demonstrated that a large proportion of the synthesized compounds selectively inhibited monoamine oxidase B with IC50 values in the sub-micromolar range. Among them, compound 5n (IC50 = 0.0601 μM) exhibited the most potent inhibitory activity and the highest selectivity for monoamine oxidase B (SI > 1664-fold). In addition, the possible binding model of the active compound 5n was measured by docking it into the active site of the hMAO-B complex structure. The results showed that compound 5n interacted with the well-known binding pocket of MAO-B, and a π–π interaction was found between the phenyl ring at position 3 of the coumarin and the phenyl ring of Tyr 326. Consequently, we supplied useful information about the interaction between the enzyme and inhibitor, and developed the 6-methyl-3-phenylcoumarin scaffold as an agent for multifaceted brain disorders.
Co-reporter:Jin-Shuai Lan, Sai-Sai Xie, Ming Huang, Ya-Jian Hu, Ling-Yi Kong and Xiao-Bing Wang
MedChemComm 2015 vol. 6(Issue 7) pp:1293-1302
Publication Date(Web):19 May 2015
DOI:10.1039/C5MD00124B
A new series of C7-substituted chromanones has been designed, synthesized and evaluated for hMAO-B inhibitory activity in vitro. Most of the studied compounds were remarkably potent and selective MAO-B inhibitors and showed weak or no inhibition of MAO-A. Especially, compound 4f (IC50 = 8.62 nM) was the best MAO-B inhibitor and exhibited the highest selectivity for MAO-B (SI > 11 627.9-fold). In addition, the structure–activity relationships for MAO-B inhibition indicated that substitutions at the C7 of the chromanone moiety, particularly with the halogen substituted benzyloxy, were more favorable for MAO-B inhibition. Molecular docking studies have been performed to explore the interaction modes of C7-substituted chromanones with MAO-B. Furthermore, the representative compounds 4f and 5d showed low neurotoxicity in SH-SY5Y cells in vitro. So the C7-substituted chromanones could be used to develop promising drug candidates for the therapy of neurodegenerative diseases.
Co-reporter:Ya-Jian Hu, Neng Jiang, Sai-Sai Xie, Su-Yi Li, Jin-Shuai Lan, Ling-Yi Kong, Xiao-Bing Wang
Tetrahedron 2015 Volume 71(Issue 42) pp:8026-8032
Publication Date(Web):21 October 2015
DOI:10.1016/j.tet.2015.08.056
An iodine-promoted convenient and environmentally friendly sequential method for the synthesis of trisubstituted methanes bearing a coumarin and a chromone ring is described. The remarkable features of this approach include avoidance of metals, working under air, employment of readily available starting materials, good functional group tolerance and simple operation.
Co-reporter:Ming Huang, Sai-Sai Xie, Neng Jiang, Jin-Shuai Lan, Ling-Yi Kong, Xiao-Bing Wang
Bioorganic & Medicinal Chemistry Letters 2015 25(3) pp: 508-513
Publication Date(Web):
DOI:10.1016/j.bmcl.2014.12.034
Co-reporter:Neng Jiang, Su-Yi Li, Sai-Sai Xie, Zhong-Rui Li, Kelvin D.G. Wang, Xiao-Bing Wang, Ling-Yi Kong
European Journal of Medicinal Chemistry 2014 Volume 87() pp:540-551
Publication Date(Web):24 November 2014
DOI:10.1016/j.ejmech.2014.10.004
•A series of salphen derivatives (1–26) were designed and synthesized.•Most of the compounds could function as antioxidants, inhibit Aβ aggregation.•Some compounds showed activity toward metal ions, neuroprotection, BBB permeability.•The metal complexes of compound 2 retained the activity of Aβ aggregation.•The metal complexes of compound 2 still showed a good neuroprotective effect.A series of salphen derivatives (1–26) have been designed, synthesized, and evaluated as chemical reagents that target and modulate multiple facets of Alzheimer's disease. Most of the compounds exhibit a significant ability to inhibit self-induced and Cu2+-induced β-amyloid (Aβ1–42) aggregation, and to function as potential antioxidants and biometal chelators. In particular, the antioxidant activity of compound 2 is 2.6-fold of the trolox value by using the ABTS radical scavenging method, and it also shows a significant ability to inhibit self-induced and Cu2+-induced β-amyloid (Aβ1–42) aggregation (70.3%, 20 μM and 85.7%, 50 μM, respectively). Moreover, it is capable of decreasing reactive oxygen species (ROS) induced by Cu2+-Aβ, shows a good neuroprotective effect in human SH-SY5Y neuroblastoma cells and can cross the blood–brain barrier. In addition, compound 2 retains the activities of antioxidant, anti Aβ aggregation and neuroprotection after capturing the metal ions Cu2+, Fe3+ and Zn2+ (its metal complexes 18, 22 and 23). Taken together, these results suggest that compound 2 might be a promising lead compound for AD treatment.Compound 2 was found to be a promising compound for further study.
Co-reporter:Jin-Shuai Lan, Sai-Sai Xie, Su-Yi Li, Long-Fei Pan, Xiao-Bing Wang, Ling-Yi Kong
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 21) pp:6089-6104
Publication Date(Web):1 November 2014
DOI:10.1016/j.bmc.2014.08.035
A series of tacrine-(β-carboline) hybrids (11a–q) were designed, synthesized and evaluated as multifunctional cholinesterase inhibitors against Alzheimer’s disease (AD). In vitro studies showed that most of them exhibited significant potency to inhibit acetylcholinesterase (eeAChE and hAChE), butyrylcholinesterase (BuChE) and self-induced β-amyloid (Aβ) aggregation, Cu2+-induced Aβ (1–42) aggregation, and to chelate metal ions. Especially, 11l presented the greatest ability to inhibit cholinesterase (IC50, 21.6 nM for eeAChE, 63.2 nM for hAChE and 39.8 nM for BuChE), good inhibition of Aβ aggregation (65.8% at 20 μM) and good antioxidant activity (1.57 trolox equivalents). Kinetic and molecular modeling studies indicated that 11l was a mixed-type inhibitor, binding simultaneously to the catalytic anionic site (CAS) and the peripheral anionic site (PAS) of AChE. In addition, 11l could chelate metal ions, reduce PC12 cells death induced by oxidative stress and penetrate the blood–brain barrier (BBB). These results suggested that 11l might be an excellent multifunctional agent for AD treatment.
Co-reporter:Neng Jiang, Su-Yi Li, Sai-Sai Xie, Hequan Yao, Hongbin Sun, Xiao-Bing Wang and Ling-Yi Kong
RSC Advances 2014 vol. 4(Issue 109) pp:63632-63641
Publication Date(Web):24 Nov 2014
DOI:10.1039/C4RA10174J
The direct intramolecular acylation of esters was developed by using the combined system of FeCl3 with Cl2CHOCH3. This unique cooperative system offered a new and efficient approach to biologically important xanthone and chromone derivatives with regioselectivity. Examples were reported, and control experiments were carried out to examine the effect of the benzyl esters and Cl2CHOCH3.

![(2S,5R,6R,7S,9R)-3,5,6,7,8,9-hexahydro-2-(2-hydroxypropan-2-yl)-6-methyl-7,9-bis(3-methylbut-2-en-1-yl)-6-(4-methylpent-3-en-1-yl)-5-(phenylcarbonyl)-5,9-methanocycloocta[b]furan-4,10(2H)-dione](http://img.cochemist.com/ccimg/1253300/1253206-39-1.png)
![(2S,5R,6R,7S,9R)-3,5,6,7,8,9-hexahydro-2-(2-hydroxypropan-2-yl)-6-methyl-7,9-bis(3-methylbut-2-en-1-yl)-6-(4-methylpent-3-en-1-yl)-5-(phenylcarbonyl)-5,9-methanocycloocta[b]furan-4,10(2H)-dione](http://img.cochemist.com/ccimg/1253300/1253206-39-1_b.png)
![(2R,5R,6R,7S,9R)-3,5,6,7,8,9-hexahydro-2-(2-hydroxypropan-2-yl)-6-methyl-7,9-bis(3-methylbut-2-en-1-yl)-6-(4-methylpent-3-en-1-yl)-5-(phenylcarbonyl)-5,9-methanocycloocta[b]furan-4,10(2H)-dione](http://img.cochemist.com/ccimg/1253300/1253206-38-0.png)
![(2R,5R,6R,7S,9R)-3,5,6,7,8,9-hexahydro-2-(2-hydroxypropan-2-yl)-6-methyl-7,9-bis(3-methylbut-2-en-1-yl)-6-(4-methylpent-3-en-1-yl)-5-(phenylcarbonyl)-5,9-methanocycloocta[b]furan-4,10(2H)-dione](http://img.cochemist.com/ccimg/1253300/1253206-38-0_b.png)
















![4,9,10-trihydroxy-2-methoxydibenzo[c,e]oxepin-5(7H)-one](http://img.cochemist.com/ccimg/1030400/1030376-89-6.png)
![4,9,10-trihydroxy-2-methoxydibenzo[c,e]oxepin-5(7H)-one](http://img.cochemist.com/ccimg/1030400/1030376-89-6_b.png)






![1-[(2-methylphenyl)methyl]piperidine-4-carboxylic Acid](http://img.cochemist.com/ccimg/897100/897094-25-6.png)
![1-[(2-methylphenyl)methyl]piperidine-4-carboxylic Acid](http://img.cochemist.com/ccimg/897100/897094-25-6_b.png)
![1H-INDOLE-3-PROPANAMIDE, N-[2-[1-(PHENYLMETHYL)-4-PIPERIDINYL]ETHYL]-](http://img.cochemist.com/ccimg/870200/870152-37-7.png)
![1H-INDOLE-3-PROPANAMIDE, N-[2-[1-(PHENYLMETHYL)-4-PIPERIDINYL]ETHYL]-](http://img.cochemist.com/ccimg/870200/870152-37-7_b.png)
![Benzaldehyde, 4-[[6-(dimethylamino)hexyl]oxy]-](http://img.cochemist.com/ccimg/855400/855342-01-7.png)
![Benzaldehyde, 4-[[6-(dimethylamino)hexyl]oxy]-](http://img.cochemist.com/ccimg/855400/855342-01-7_b.png)


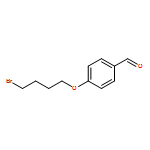
![Benzaldehyde, 4-[(7-bromoheptyl)oxy]-](http://img.cochemist.com/ccimg/482400/482359-35-3.png)
![Benzaldehyde, 4-[(7-bromoheptyl)oxy]-](http://img.cochemist.com/ccimg/482400/482359-35-3_b.png)
![1H-Inden-1-one,2,3-dihydro-5,6-dimethoxy-2-[[1-(2-pyridinylmethyl)-4-piperidinyl]methyl]-](http://img.cochemist.com/ccimg/388200/388115-22-8.png)
![1H-Inden-1-one,2,3-dihydro-5,6-dimethoxy-2-[[1-(2-pyridinylmethyl)-4-piperidinyl]methyl]-](http://img.cochemist.com/ccimg/388200/388115-22-8_b.png)




![Phenol, 2-[(E)-[(4-hydroxyphenyl)imino]methyl]-](http://img.cochemist.com/ccimg/237500/237418-42-7.png)
![Phenol, 2-[(E)-[(4-hydroxyphenyl)imino]methyl]-](http://img.cochemist.com/ccimg/237500/237418-42-7_b.png)


![1H-Pyrido[3,4-b]indole-3-carboxylic acid, 2,3,4,9-tetrahydro-1-phenyl-, (3S)-](/data/chemimg/1787000/209169-30-2.png)
![1H-Pyrido[3,4-b]indole-3-carboxylic acid, 2,3,4,9-tetrahydro-1-phenyl-, (3S)-](/data/chemimg/1787000/209169-30-2_b.png)


![1H-Pyrido[3,4-b]indole-3-carboxylic acid, 1-ethyl-2,3,4,9-tetrahydro-, (3S)-](/data/chemimg/1776200/191279-45-5.png)
![1H-Pyrido[3,4-b]indole-3-carboxylic acid, 1-ethyl-2,3,4,9-tetrahydro-, (3S)-](/data/chemimg/1776200/191279-45-5_b.png)
![1H-Pyrido[3,4-b]indole-3-carboxylic acid, 2,3,4,9-tetrahydro-1-methyl-, (3S)-](/data/chemimg/1776500/191279-37-5.png)
![1H-Pyrido[3,4-b]indole-3-carboxylic acid, 2,3,4,9-tetrahydro-1-methyl-, (3S)-](/data/chemimg/1776500/191279-37-5_b.png)
![4-PIPERIDINEACETIC ACID, 1-[(4-CHLOROPHENYL)METHYL]-, ETHYL ESTER](http://img.cochemist.com/ccimg/185500/185434-41-7.png)
![4-PIPERIDINEACETIC ACID, 1-[(4-CHLOROPHENYL)METHYL]-, ETHYL ESTER](http://img.cochemist.com/ccimg/185500/185434-41-7_b.png)



![Benzaldehyde, 4-[(8-bromooctyl)oxy]-](http://img.cochemist.com/ccimg/165600/165550-59-4.png)
![Benzaldehyde, 4-[(8-bromooctyl)oxy]-](http://img.cochemist.com/ccimg/165600/165550-59-4_b.png)






![Benzaldehyde, 4-[(5-bromopentyl)oxy]-](http://img.cochemist.com/ccimg/143800/143773-71-1.png)
![Benzaldehyde, 4-[(5-bromopentyl)oxy]-](http://img.cochemist.com/ccimg/143800/143773-71-1_b.png)




![Benzaldehyde, 4-[[6-(diethylamino)hexyl]oxy]-](http://img.cochemist.com/ccimg/135000/134994-20-0.png)
![Benzaldehyde, 4-[[6-(diethylamino)hexyl]oxy]-](http://img.cochemist.com/ccimg/135000/134994-20-0_b.png)


![(3E)-3-[(4-hydroxy-3-methoxyphenyl)methylidene]-2,3-dihydro-4H-chromen-4-one](http://img.cochemist.com/ccimg/132800/132794-01-5.png)
![(3E)-3-[(4-hydroxy-3-methoxyphenyl)methylidene]-2,3-dihydro-4H-chromen-4-one](http://img.cochemist.com/ccimg/132800/132794-01-5_b.png)


![4-PYRIDINECARBOXAMIDE, N-[2-[1-(PHENYLMETHYL)-4-PIPERIDINYL]ETHYL]-](http://img.cochemist.com/ccimg/126900/126848-98-4.png)
![4-PYRIDINECARBOXAMIDE, N-[2-[1-(PHENYLMETHYL)-4-PIPERIDINYL]ETHYL]-](http://img.cochemist.com/ccimg/126900/126848-98-4_b.png)
![Benzamide, 4-methoxy-N-[2-[1-(phenylmethyl)-4-piperidinyl]ethyl]-](http://img.cochemist.com/ccimg/126900/126848-89-3.png)
![Benzamide, 4-methoxy-N-[2-[1-(phenylmethyl)-4-piperidinyl]ethyl]-](http://img.cochemist.com/ccimg/126900/126848-89-3_b.png)
![Benzamide, 3-nitro-N-[2-[1-(phenylmethyl)-4-piperidinyl]ethyl]-](http://img.cochemist.com/ccimg/126900/126848-87-1.png)
![Benzamide, 3-nitro-N-[2-[1-(phenylmethyl)-4-piperidinyl]ethyl]-](http://img.cochemist.com/ccimg/126900/126848-87-1_b.png)
![Benzamide, 4-methyl-N-[2-[1-(phenylmethyl)-4-piperidinyl]ethyl]-](http://img.cochemist.com/ccimg/126900/126848-83-7.png)
![Benzamide, 4-methyl-N-[2-[1-(phenylmethyl)-4-piperidinyl]ethyl]-](http://img.cochemist.com/ccimg/126900/126848-83-7_b.png)
![3H-Pyrano[3',4':6,7]indolizino[1,2-b]quinoline-11,14-dione,4-ethenyl-3-(b-D-glucopyranosyloxy)-4,4a,5,5a,6,12-hexahydro-,(3S,4R,4aS,5aS)- (9CI)](http://img.cochemist.com/ccimg/126800/126722-26-7.png)
![3H-Pyrano[3',4':6,7]indolizino[1,2-b]quinoline-11,14-dione,4-ethenyl-3-(b-D-glucopyranosyloxy)-4,4a,5,5a,6,12-hexahydro-,(3S,4R,4aS,5aS)- (9CI)](http://img.cochemist.com/ccimg/126800/126722-26-7_b.png)
![(3S-(3alpha,4beta,4aalpha,5abeta))-4-Ethenyl-3-(beta-D-glucopyranosyloxy)-4,4a,5,5a,6,12-hexahydro-3H-pyrano[3',4':6,7]indolizino[1,2-b]quinoline-11,14-dione](http://img.cochemist.com/ccimg/126700/126624-21-3.png)
![(3S-(3alpha,4beta,4aalpha,5abeta))-4-Ethenyl-3-(beta-D-glucopyranosyloxy)-4,4a,5,5a,6,12-hexahydro-3H-pyrano[3',4':6,7]indolizino[1,2-b]quinoline-11,14-dione](http://img.cochemist.com/ccimg/126700/126624-21-3_b.png)






![9H-Pyrido[3,4-b]indole-3-carboxylic acid, 1-(4-methoxyphenyl)-, methyl ester](/data/chemimg/3705000/113247-18-0.png)
![9H-Pyrido[3,4-b]indole-3-carboxylic acid, 1-(4-methoxyphenyl)-, methyl ester](/data/chemimg/3705000/113247-18-0_b.png)
![3-Pyridinecarboxamide, N-[2-[1-(phenylmethyl)-4-piperidinyl]ethyl]-](http://img.cochemist.com/ccimg/113100/113027-62-6.png)
![3-Pyridinecarboxamide, N-[2-[1-(phenylmethyl)-4-piperidinyl]ethyl]-](http://img.cochemist.com/ccimg/113100/113027-62-6_b.png)
![Benzamide, 4-chloro-N-[2-[1-(phenylmethyl)-4-piperidinyl]ethyl]-](http://img.cochemist.com/ccimg/113100/113027-44-4.png)
![Benzamide, 4-chloro-N-[2-[1-(phenylmethyl)-4-piperidinyl]ethyl]-](http://img.cochemist.com/ccimg/113100/113027-44-4_b.png)
![Benzamide, 4-nitro-N-[2-[1-(phenylmethyl)-4-piperidinyl]ethyl]-](http://img.cochemist.com/ccimg/113100/113027-40-0.png)
![Benzamide, 4-nitro-N-[2-[1-(phenylmethyl)-4-piperidinyl]ethyl]-](http://img.cochemist.com/ccimg/113100/113027-40-0_b.png)






![Phenol, 2-[(E)-(phenylimino)methyl]-](http://img.cochemist.com/ccimg/103900/103886-92-6.png)
![Phenol, 2-[(E)-(phenylimino)methyl]-](http://img.cochemist.com/ccimg/103900/103886-92-6_b.png)








![9H-Pyrido[3,4-b]indole-3-carboxylic acid, 1-phenyl-, methyl ester](/data/chemimg/1637000/81677-50-1.png)
![9H-Pyrido[3,4-b]indole-3-carboxylic acid, 1-phenyl-, methyl ester](/data/chemimg/1637000/81677-50-1_b.png)
![1H-Pyrido[3,4-b]indole-3-carboxylic acid, 2,3,4,9-tetrahydro-, methyl ester, (3S)-](/data/chemimg/704000/79815-18-2.png)
![1H-Pyrido[3,4-b]indole-3-carboxylic acid, 2,3,4,9-tetrahydro-, methyl ester, (3S)-](/data/chemimg/704000/79815-18-2_b.png)


![9H-Pyrido[3,4-b]indole-3-carboxylic acid, 1-ethyl-, methyl ester](/data/chemimg/693100/75304-04-0.png)
![9H-Pyrido[3,4-b]indole-3-carboxylic acid, 1-ethyl-, methyl ester](/data/chemimg/693100/75304-04-0_b.png)


![9H-PYRIDO[3,4-B]INDOLE-3-CARBOXYLIC ACID](http://img.cochemist.com/ccimg/74300/74214-63-4.png)
![9H-PYRIDO[3,4-B]INDOLE-3-CARBOXYLIC ACID](http://img.cochemist.com/ccimg/74300/74214-63-4_b.png)



![9H-Pyrido[3,4-b]indole-3-carboxylicacid, methyl ester](http://img.cochemist.com/ccimg/70000/69954-48-9.png)
![9H-Pyrido[3,4-b]indole-3-carboxylicacid, methyl ester](http://img.cochemist.com/ccimg/70000/69954-48-9_b.png)
![(4S)-4-ethyl-4-hydroxy-6,12-dihydro-1H-pyrano[3',4':6,7]indolizino[1,2-b]quinoline-3,8,14(4H)-trione](http://img.cochemist.com/ccimg/68500/68426-53-9.png)
![(4S)-4-ethyl-4-hydroxy-6,12-dihydro-1H-pyrano[3',4':6,7]indolizino[1,2-b]quinoline-3,8,14(4H)-trione](http://img.cochemist.com/ccimg/68500/68426-53-9_b.png)







![7H-benzo[c]xanthen-7-one](http://img.cochemist.com/ccimg/63200/63154-69-8.png)
![7H-benzo[c]xanthen-7-one](http://img.cochemist.com/ccimg/63200/63154-69-8_b.png)















![9-chloro-2,3-dihydro-1H-cyclopenta[b]quinoline](http://img.cochemist.com/ccimg/40600/40528-00-5.png)
![9-chloro-2,3-dihydro-1H-cyclopenta[b]quinoline](http://img.cochemist.com/ccimg/40600/40528-00-5_b.png)

![8H-6,11b-Epoxy-4b,8:7a,10-dimethano-1H-2-benzopyrano[5,6-d][1,3]benzodioxepin-11-acetic acid, 1-(3-furanyl)decahydro-8,9,16-trihydroxy-6,10,11a,13a-tetramethyl-3-oxo-, methyl ester, (1R,4aR,4bR,6S,7aR,8S,9S,10R,11S,11aR,11bS,13aR,16R)-](/data/chemimg/3771000/35183-64-3.png)
![8H-6,11b-Epoxy-4b,8:7a,10-dimethano-1H-2-benzopyrano[5,6-d][1,3]benzodioxepin-11-acetic acid, 1-(3-furanyl)decahydro-8,9,16-trihydroxy-6,10,11a,13a-tetramethyl-3-oxo-, methyl ester, (1R,4aR,4bR,6S,7aR,8S,9S,10R,11S,11aR,11bS,13aR,16R)-](/data/chemimg/3771000/35183-64-3_b.png)


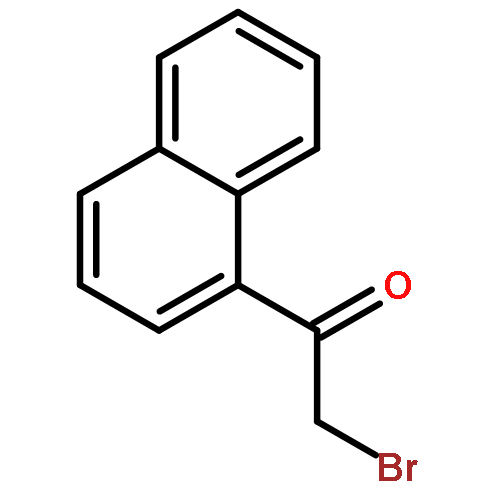
![2-BROMO-1-[4-(2-METHYLPROPYL)PHENYL]ETHANONE](http://img.cochemist.com/ccimg/30100/30095-48-8.png)
![2-BROMO-1-[4-(2-METHYLPROPYL)PHENYL]ETHANONE](http://img.cochemist.com/ccimg/30100/30095-48-8_b.png)
![6H-Dibenzo[b,d]pyran-6-one,3,7-dihydroxy-9-methoxy-1-methyl-](http://img.cochemist.com/ccimg/23500/23452-05-3.png)
![6H-Dibenzo[b,d]pyran-6-one,3,7-dihydroxy-9-methoxy-1-methyl-](http://img.cochemist.com/ccimg/23500/23452-05-3_b.png)
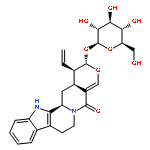

![5H-Indolo[2,3-a]pyrano[3,4-g]quinolizin-5-one,1-ethenyl-2-(b-D-glucopyranosyloxy)-1,2,7,8,13,13b,14,14a-octahydro-,(1R,2S,13bS,14aS)-](http://img.cochemist.com/ccimg/23200/23141-25-5.png)
![5H-Indolo[2,3-a]pyrano[3,4-g]quinolizin-5-one,1-ethenyl-2-(b-D-glucopyranosyloxy)-1,2,7,8,13,13b,14,14a-octahydro-,(1R,2S,13bS,14aS)-](http://img.cochemist.com/ccimg/23200/23141-25-5_b.png)
![9H-Pyrido[3,4-b]indole-3-carboxylicacid, 1-methyl-](http://img.cochemist.com/ccimg/22400/22329-38-0.png)
![9H-Pyrido[3,4-b]indole-3-carboxylicacid, 1-methyl-](http://img.cochemist.com/ccimg/22400/22329-38-0_b.png)
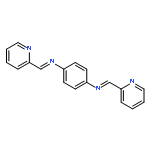









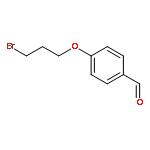
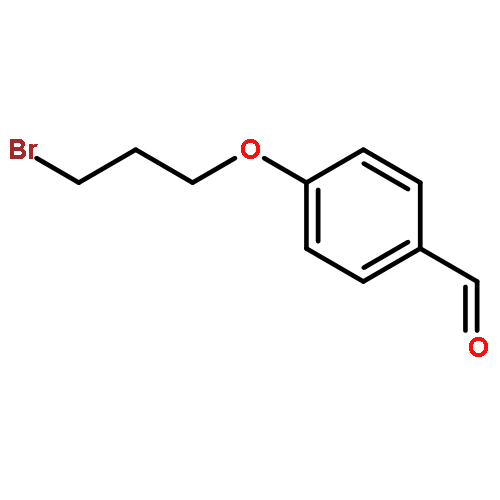
![Phenol, 2,2'-[1,3-phenylenebis(nitrilomethylidyne)]bis-](/data/chemimg/1766500/17911-94-3.png)
![Phenol, 2,2'-[1,3-phenylenebis(nitrilomethylidyne)]bis-](/data/chemimg/1766500/17911-94-3_b.png)


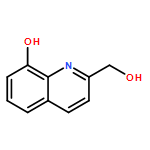
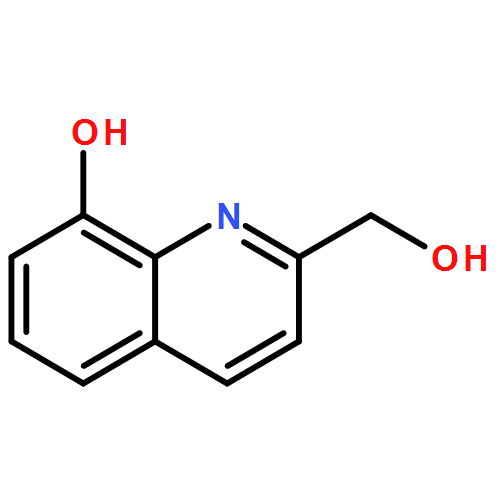
![9H-Pyrido[3,4-b]indole-3-carboxylic acid, 1-methyl-, methyl ester](/data/chemimg/924600/16641-82-0.png)
![9H-Pyrido[3,4-b]indole-3-carboxylic acid, 1-methyl-, methyl ester](/data/chemimg/924600/16641-82-0_b.png)
![Phenol, 4-chloro-2-[(E)-(phenylimino)methyl]-](http://img.cochemist.com/ccimg/15600/15501-17-4.png)
![Phenol, 4-chloro-2-[(E)-(phenylimino)methyl]-](http://img.cochemist.com/ccimg/15600/15501-17-4_b.png)
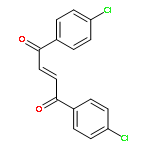
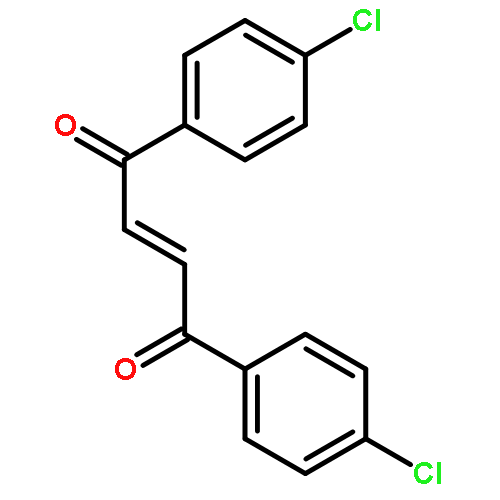
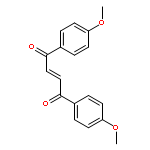
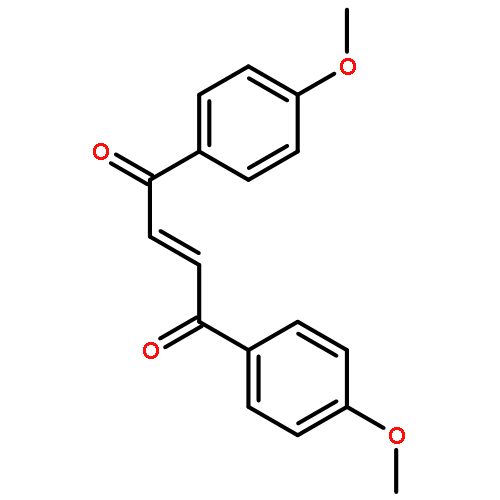

![Bicyclo[7.2.0]undec-4-ene,4,11,11-trimethyl-8-methylene-](http://img.cochemist.com/ccimg/13900/13877-93-5.png)
![Bicyclo[7.2.0]undec-4-ene,4,11,11-trimethyl-8-methylene-](http://img.cochemist.com/ccimg/13900/13877-93-5_b.png)



![Phenol,2,2'-[1,4-phenylenebis(methylidynenitrilo)]bis-](http://img.cochemist.com/ccimg/13100/13060-68-9.png)
![Phenol,2,2'-[1,4-phenylenebis(methylidynenitrilo)]bis-](http://img.cochemist.com/ccimg/13100/13060-68-9_b.png)
![N-[2-(3,4-dimethoxyphenyl)ethyl]-2-(4-hydroxyphenyl)acetamide](http://img.cochemist.com/ccimg/10300/10214-84-3.png)
![N-[2-(3,4-dimethoxyphenyl)ethyl]-2-(4-hydroxyphenyl)acetamide](http://img.cochemist.com/ccimg/10300/10214-84-3_b.png)


![BENZALDEHYDE, 4-[2-[METHYL(PHENYLMETHYL)AMINO]ETHOXY]-](http://img.cochemist.com/ccimg/7400/7319-57-5.png)
![BENZALDEHYDE, 4-[2-[METHYL(PHENYLMETHYL)AMINO]ETHOXY]-](http://img.cochemist.com/ccimg/7400/7319-57-5_b.png)



![11-chloro-7,8,9,10-tetrahydro-6H-cyclohepta[b]quinoline](http://img.cochemist.com/ccimg/5800/5778-71-2.png)
![11-chloro-7,8,9,10-tetrahydro-6H-cyclohepta[b]quinoline](http://img.cochemist.com/ccimg/5800/5778-71-2_b.png)


![6-[[4-[(6-oxocyclohexa-2,4-dien-1-ylidene)methylamino]anilino]methylidene]cyclohexa-2,4-dien-1-one](http://img.cochemist.com/ccimg/5400/5344-68-3.png)
![6-[[4-[(6-oxocyclohexa-2,4-dien-1-ylidene)methylamino]anilino]methylidene]cyclohexa-2,4-dien-1-one](http://img.cochemist.com/ccimg/5400/5344-68-3_b.png)


![Phenol, 2-[(E)-[(2-hydroxyphenyl)imino]methyl]-](http://img.cochemist.com/ccimg/1700/1624-54-0.png)
![Phenol, 2-[(E)-[(2-hydroxyphenyl)imino]methyl]-](http://img.cochemist.com/ccimg/1700/1624-54-0_b.png)
![Phenol, 2-[(E)-(phenylmethylene)amino]-](http://img.cochemist.com/ccimg/1700/1624-53-9.png)
![Phenol, 2-[(E)-(phenylmethylene)amino]-](http://img.cochemist.com/ccimg/1700/1624-53-9_b.png)

![Benzamide,N-[1-(phenylmethyl)-4-piperidinyl]-](http://img.cochemist.com/ccimg/1000/971-34-6.png)
![Benzamide,N-[1-(phenylmethyl)-4-piperidinyl]-](http://img.cochemist.com/ccimg/1000/971-34-6_b.png)
![6H-Dibenzo[b,d]pyran-6-one,3,7,9-trihydroxy-1-methyl-](http://img.cochemist.com/ccimg/700/641-38-3.png)
![6H-Dibenzo[b,d]pyran-6-one,3,7,9-trihydroxy-1-methyl-](http://img.cochemist.com/ccimg/700/641-38-3_b.png)







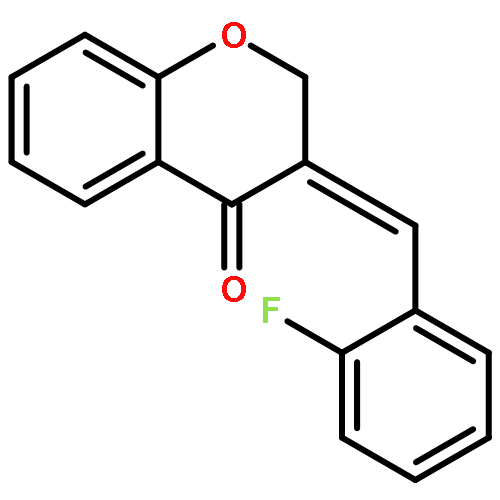


![1-{4-[(3-Methylbenzyl)oxy]phenyl}ethanone](http://img.cochemist.com/ccimg/890100/890092-06-5.png)
![1-{4-[(3-Methylbenzyl)oxy]phenyl}ethanone](http://img.cochemist.com/ccimg/890100/890092-06-5_b.png)
![1-{3-[(4-nitrobenzyl)oxy]phenyl}ethanone](http://img.cochemist.com/ccimg/694500/694455-76-0.png)
![1-{3-[(4-nitrobenzyl)oxy]phenyl}ethanone](http://img.cochemist.com/ccimg/694500/694455-76-0_b.png)
![1-{3-[(3-nitrobenzyl)oxy]phenyl}ethanone](http://img.cochemist.com/ccimg/694500/694451-42-8.png)
![1-{3-[(3-nitrobenzyl)oxy]phenyl}ethanone](http://img.cochemist.com/ccimg/694500/694451-42-8_b.png)
![ETHANONE, 1-[4-[[2-(TRIFLUOROMETHYL)PHENYL]METHOXY]PHENYL]-](http://img.cochemist.com/ccimg/478200/478163-22-3.png)
![ETHANONE, 1-[4-[[2-(TRIFLUOROMETHYL)PHENYL]METHOXY]PHENYL]-](http://img.cochemist.com/ccimg/478200/478163-22-3_b.png)
![2H-1-Benzopyran-2-one, 7-[(3,4-difluorophenyl)methoxy]-3,4-dimethyl-](http://img.cochemist.com/ccimg/320400/320350-94-5.png)
![2H-1-Benzopyran-2-one, 7-[(3,4-difluorophenyl)methoxy]-3,4-dimethyl-](http://img.cochemist.com/ccimg/320400/320350-94-5_b.png)


![Ethanone, 1-[3-[(3-chlorophenyl)methoxy]phenyl]-](http://img.cochemist.com/ccimg/206700/206653-46-5.png)
![Ethanone, 1-[3-[(3-chlorophenyl)methoxy]phenyl]-](http://img.cochemist.com/ccimg/206700/206653-46-5_b.png)
![Ethanone, 1-[4-[(2,4-difluorophenyl)methoxy]phenyl]-](http://img.cochemist.com/ccimg/187600/187532-79-2.png)
![Ethanone, 1-[4-[(2,4-difluorophenyl)methoxy]phenyl]-](http://img.cochemist.com/ccimg/187600/187532-79-2_b.png)
![Ethanone, 1-[3-[(4-fluorophenyl)methoxy]phenyl]-](http://img.cochemist.com/ccimg/144400/144337-72-4.png)
![Ethanone, 1-[3-[(4-fluorophenyl)methoxy]phenyl]-](http://img.cochemist.com/ccimg/144400/144337-72-4_b.png)


![4H-1-Benzopyran-4-one, 2,3-dihydro-3-[[4-(1-methylethyl)phenyl]methylene]-, (E)-](http://img.cochemist.com/ccimg/109800/109754-23-6.png)
![4H-1-Benzopyran-4-one, 2,3-dihydro-3-[[4-(1-methylethyl)phenyl]methylene]-, (E)-](http://img.cochemist.com/ccimg/109800/109754-23-6_b.png)
![Ethanone, 1-[4-[(3-nitrophenyl)methoxy]phenyl]-](http://img.cochemist.com/ccimg/104000/103981-34-6.png)
![Ethanone, 1-[4-[(3-nitrophenyl)methoxy]phenyl]-](http://img.cochemist.com/ccimg/104000/103981-34-6_b.png)


![1-{3-[(4-chlorobenzyl)oxy]phenyl}ethanone](http://img.cochemist.com/ccimg/79700/79615-79-5.png)
![1-{3-[(4-chlorobenzyl)oxy]phenyl}ethanone](http://img.cochemist.com/ccimg/79700/79615-79-5_b.png)
![1-[4-[(4-METHYLPHENYL)METHOXY]PHENYL]ETHANONE](http://img.cochemist.com/ccimg/79700/79615-78-4.png)
![1-[4-[(4-METHYLPHENYL)METHOXY]PHENYL]ETHANONE](http://img.cochemist.com/ccimg/79700/79615-78-4_b.png)
![Ethanone, 1-[4-[[3-(trifluoromethyl)phenyl]methoxy]phenyl]-](http://img.cochemist.com/ccimg/79700/79615-77-3.png)
![Ethanone, 1-[4-[[3-(trifluoromethyl)phenyl]methoxy]phenyl]-](http://img.cochemist.com/ccimg/79700/79615-77-3_b.png)
![ETHANONE, 1-[4-[(3-CHLOROPHENYL)METHOXY]PHENYL]-](http://img.cochemist.com/ccimg/79700/79615-76-2.png)
![ETHANONE, 1-[4-[(3-CHLOROPHENYL)METHOXY]PHENYL]-](http://img.cochemist.com/ccimg/79700/79615-76-2_b.png)
![1-[4-[(2-CHLOROPHENYL)METHOXY]PHENYL]ETHANONE](http://img.cochemist.com/ccimg/72300/72293-95-9.png)
![1-[4-[(2-CHLOROPHENYL)METHOXY]PHENYL]ETHANONE](http://img.cochemist.com/ccimg/72300/72293-95-9_b.png)
![1-{4-[(2-Methylbenzyl)oxy]phenyl}ethanone](http://img.cochemist.com/ccimg/72300/72293-94-8.png)
![1-{4-[(2-Methylbenzyl)oxy]phenyl}ethanone](http://img.cochemist.com/ccimg/72300/72293-94-8_b.png)




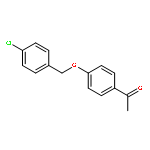

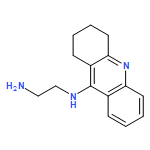
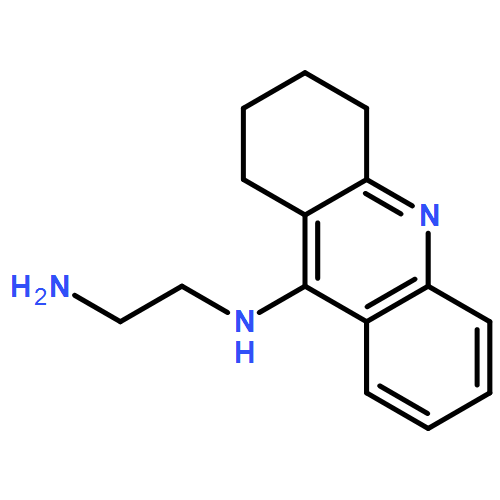
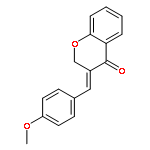

![6H-Dibenzo[b,d]pyran-6-one,2,3,4,4a-tetrahydro-2,3,7-trihydroxy-9-methoxy-4a-methyl-, (2R,3R,4aR)-rel-](http://img.cochemist.com/ccimg/29800/29752-43-0.png)
![6H-Dibenzo[b,d]pyran-6-one,2,3,4,4a-tetrahydro-2,3,7-trihydroxy-9-methoxy-4a-methyl-, (2R,3R,4aR)-rel-](http://img.cochemist.com/ccimg/29800/29752-43-0_b.png)