Co-reporter:Lei Yu, Minhao Huang, Tianfeng Xu, Linjiang Tong, Xiao-e Yan, Zhang Zhang, Yong Xu, Caihong Yun, Hua Xie, Ke Ding, Xiaoyun Lu
European Journal of Medicinal Chemistry 2017 Volume 126(Volume 126) pp:
Publication Date(Web):27 January 2017
DOI:10.1016/j.ejmech.2016.12.006
•Optimization of pyrido[2,3-d]pyrimidin-7-ones with improved pharmacokinetic properties.•Compound 9s potently suppressed EGFRL858R/T790M kinase and H1975 cells.•Compound 9s exhibited moderate plasma exposure and an oral bioavailability value of 16%.Structural optimization of pyrido[2,3-d]pyrimidin-7-ones was conducted to yield a series of new selective EGFRT790M inhibitors with improved pharmacokinetic properties. One of the most promising compound 9s potently suppressed EGFRL858R/T790M kinase and inhibited the proliferation of H1975 cells with IC50 values of 2.0 nM and 40 nM, respectively. The compound dose-dependently induced reduction of the phosphorylation of EGFR and downstream activation of ERK in NCIH1975 cells. It also exhibited moderate plasma exposure after oral administration and an oral bioavailability value of 16%. Compound 9s may serve as a promising lead compound for further drug discovery overcoming the acquired resistance of non-small cell lung cancer (NSCLC) patients.Structural optimization of pyrido[2,3-d]pyrimidin-7-ones yielded new selective EGFRT790M inhibitors. Compound 9s exhibited good pharmacokinetic properties with F value of 16%, and inhibited EGFRL858R/T790M kinase and H1975 cells with IC50 values of 2.0 and 40 nM, respectively.Download high-res image (191KB)Download full-size image
Co-reporter:Wei Yan;Zhaoru Huang;Zhengyu Wang;Sufen Cao;Linjiang Tong;Tao Zhang;Chen Wang;Lin Zhou;Jian Ding;Cheng Luo;Jinpei Zhou;Wenhu Duan
Chemical Biology & Drug Design 2016 Volume 87( Issue 5) pp:694-703
Publication Date(Web):
DOI:10.1111/cbdd.12703
In this study, we described the design, synthesis, and biological evaluation of 1,3-diaryl-pyridones as vascular endothelial growth factor receptor-2 (VEGFR-2) inhibitors. The 1,3-diaryl-pyridones were synthesized via Chan-Lam and Suzuki coupling reactions. Two representative compounds, 17 and 35h, displayed excellent enzymatic inhibitory activities, with IC50 values of 3.5 and 3.0 nm, respectively. Furthermore, compounds 17 and 35h blocked the tube formation and suppressed the VEGF-induced phosphorylation of VEGFR-2 and downstream extracellular signal-regulated kinases (Erk) in human umbilical vein endothelial cells (HUVECs) at 10 nm concentration. The docking simulation showed that compound 17 bound well into the active site of VEGFR-2 via two hydrogen bonds and hydrophobic interactions.
Co-reporter:Qiang Xiao, Rong Qu, Dingding Gao, Qi Yan, Linjiang Tong, Wei Zhang, Jian Ding, Hua Xie, Yingxia Li
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 12) pp:2673-2680
Publication Date(Web):15 June 2016
DOI:10.1016/j.bmc.2016.04.032
To overcome the drug-resistance of first generation EGFR inhibitors and the nonselective toxicities of second generation inhibitors among NSCLC patients, a series of 5-(methylthio)pyrimidine derivatives were discovered as novel EGFR inhibitors, which harbored not only potent enzymatic and antiproliferative activities against EGFRL858R/T790M mutants, but good selectivity over wide-type form of the receptor. This goal was achieved by employing structure-based drug design and traditional optimization strategies, based on WZ4002 and CO1686. These derivatives inhibited the enzymatic activity of EGFRL858R/T790M mutants with IC50 values in subnanomolar ranges, while exhibiting hundreds of fold less potency on EGFRWT. These compounds also strongly inhibited the proliferation of H1975 non-small cell lung cancer cells bearing EGFRL858R/T790M, while being significantly less toxic to A431 human epithelial carcinoma cells with overexpressed EGFRWT. The EGFR kinase inhibitory and antiproliferative activities were further validated by Western blot analysis for activation of EGFR and the downstream signaling in cancer cells.
Co-reporter:Guo-Rui Gao, Meng-Yuan Li, Lin-Jiang Tong, Li-Xin Wei, Jian Ding, Hua Xie, Wen-Hu Duan
Chinese Chemical Letters 2015 Volume 26(Issue 9) pp:1165-1168
Publication Date(Web):September 2015
DOI:10.1016/j.cclet.2015.07.016
Inhibition of VEGFR-2 signaling pathway has already become one of the most promising approaches for the treatment of cancer. In this study, we describe the design, synthesis, and biological evaluation of a series of O-linked indoles as potent inhibitors of VEGFR-2. Among these compounds, 18 showed significant anti-angiogenesis activities via VEGFR-2 in enzymatic proliferation assays, with IC50 value of 3.8 nmol/L. Kinase selectivity profiling revealed that 18 was a multitargeted inhibitor, and it also exhibited good potency against VEGFR-1, PDGFR-α and β.In an effort to discover potent VEGFR-2 inhibitors, a series of 2,4 or 4,6-disubstituted O-linked indoles derivatives were designed and synthesized. The structural activity relationships led to identification of a potential VEGFR-2 inhibitor compound 18.
Co-reporter:Shingpan Chan, Kun Han, Rong Qu, Linjiang Tong, Yingjun Li, Zhang Zhang, Huimin Cheng, Xiaoyun Lu, Adam Patterson, Jeff Smaill, Xiaomei Ren, Jian Ding, Hua Xie, Ke Ding
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 19) pp:4277-4281
Publication Date(Web):1 October 2015
DOI:10.1016/j.bmcl.2015.07.089
IGF1R amplification was recently implied to be related to the secondary acquired resistance against the 2nd or 3rd generation EGFR inhibitor therapies. We have successfully identified a series of 2,4-diarylamino-pyrimidines as new IGF1R/EGFRL858R/T790M co-targeting agents. One of the most promising compounds 8g potently inhibits both kinases with low nanomolar IC50 values, but is significantly less potent in inhibiting the wild type EGFR. The compound also displays a good kinase selectivity profile against a panel of 468 kinases. Moreover, 8g strongly suppresses the proliferation of CO-1686-resistant H1975-IGF1R cancer cells, suggesting its promising potential as a new lead compound for future anticancer drug discovery.
Co-reporter:Shaoying Tan, Kun Han, Qiang Li, Linjiang Tong, Yiqi Yang, Zhuo Chen, Hua Xie, Jian Ding, Xuhong Qian, Yufang Xu
European Journal of Medicinal Chemistry 2014 Volume 85() pp:207-214
Publication Date(Web):6 October 2014
DOI:10.1016/j.ejmech.2014.07.068
•Novel metal complexes of naphthalimide–cyclam conjugates were designed, synthesized.•All of the compounds were evaluated of their antitumor activities.•Zn(II) and Cr(III) complexes displayed as multi-target RTK inhibitors.•Representative compound 8a showed antiproliferative and antiangiogenic activities.A novel series of metal complexes of naphthalimide–cyclam conjugates were synthesized and their in vitro antitumor activities were evaluated. The newly-synthesized compounds showed huge diversity of antiproliferative potency due to variety of metal ions and length of alkyl chains, among which the Zn(II) and Cr(III) complexes exhibited comparable antiproliferative activities with amonafide via multiple tyrosine kinase inhibition. Further research revealed that the representative compound 8a displayed broad-spectrum antiproliferative activity against 15 cancer cell lines with average IC50 value 10.18 ± 3.25 μM, and effective antiangiogenic activity on human microvascular endothelial cells (HMEC-1). In brief, metal complexes of naphthalimide–cyclam conjugates were firstly designed and synthesized as multi-target tyrosine kinase inhibitors and proved of their antitumor capacities.A novel series of metal complexes of naphthalimide–cyclam conjugates were designed and synthesized, which displayed as multi-target RTK inhibitors. Representative compound 8a exhibited both broad-spectrum antiproliferative capacity and antiangiogenic activity.
Co-reporter:Xun Ji, Ting Peng, Xu Zhang, Jian Li, Wei Yang, Linjiang Tong, Rong Qu, Hualiang Jiang, Jian Ding, Hua Xie, Hong Liu
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 7) pp:2366-2378
Publication Date(Web):1 April 2014
DOI:10.1016/j.bmc.2014.01.035
A novel series of 6-alkenylamides of 4-anilinothieno[2,3-d]pyrimidine derivatives was designed, synthesized and evaluated as irreversible inhibitors of the epidermal growth factor receptor (EGFR). Most of the compounds exhibited good potency against EGFR wild type (EGFR wt) and EGFR T790M/L858R. Among these, the half-maximal inhibitory concentration (IC50) values of 17 compounds against EGFR wt were less than 0.020 μM, and those of 12 compounds were less than 0.010 μM. The IC50 values of 10 compounds against EGFR T790M/L858R were less than 0.005 μM. Compounds 8l, 9n, 9o, 9q and 9v almost completely blocked the phosphorylation of EGFR in the A431 cell line at 1 μM. Compounds 8l, 9n, 9o, 9q and 9v blocked the autophosphorylation of EGFR in NCI-H1975 cells at high concentration (1 μM), and compound 8l was confirmed to be an irreversible inhibitor through the dilution method.
Co-reporter:Yongcong Lv, Mengyuan Li, Ting Liu, Linjiang Tong, Ting Peng, Lixin Wei, Jian Ding, Hua Xie, and Wenhu Duan
ACS Medicinal Chemistry Letters 2014 Volume 5(Issue 5) pp:592-597
Publication Date(Web):February 24, 2014
DOI:10.1021/ml5000417
Inhibition of VEGFR-2 signaling pathway has already become one of the most promising approaches for the treatment of cancer. In this study, we describe the design, synthesis, and biological evaluation of a new series of naphthamides as potent inhibitors of VEGFR-2. Among these compounds, 14c exhibited high VEGFR-2 inhibitory potency in both enzymatic and HUVEC cellular proliferation assays, with IC50 values of 1.5 and 0.9 nM, respectively. Kinase selectivity profiling revealed that 14c was a multitargeted inhibitor, and it also exhibited good potency against VEGFR-1, PDGFR-β, and RET. Furthermore, 14c effectively blocked tube formation of HUVEC at nanomolar level. Overall, 14c might be a promising candidate for the treatment of cancer.Keywords: Angiogenesis; HUVEC; inhibitor; naphthamide; VEGFR-2;
Co-reporter:Li-Ping Liu, Kun Han, Wei Chen, Ying-Ying Zhang, Lin-Jiang Tong, Ting Peng, Hua Xie, Jian Ding, Hong-Bing Wang
Bioorganic & Medicinal Chemistry 2014 22(15) pp: 4198-4203
Publication Date(Web):
DOI:10.1016/j.bmc.2014.05.042
Co-reporter:Xin Wang, Zhuo Chen, Linjiang Tong, Shaoying Tan, Wei Zhou, Ting Peng, Kun Han, Jian Ding, Hua Xie, Yufang Xu
European Journal of Medicinal Chemistry 2013 Volume 65() pp:477-486
Publication Date(Web):July 2013
DOI:10.1016/j.ejmech.2013.05.002
•Novel naphthalimide derivatives were synthesized.•The compounds were topo II inhibitors and showed good antiproliferative activity.•For the first time, naphthalimides were proved to be tyrosine kinase inhibitors.•8d displayed effective antiangiogentic activity by inhibiting tyrosine kinases.Novel naphthalimide derivatives were designed and synthesized to modulate both topoisomerase II (topo II) and receptor tyrosine kinases (RTKs). Most target compounds exhibited effective and selective antiproliferative activities against three cancer cell lines by inhibiting topo II. The IC50 values ranged from 1.5 to 19.1 μM. Moreover, compounds 8d and 12d moderately inhibited various angiogenesis-related RTKs, including FGFR1, VEGFR2 and PDGFRα. The representative compound 8d was then proved to possess antiangiogenic activity, which was evidenced by the inhibition of migration and tube formation activities of HMEC-1 cells. To our knowledge, it is the first time naphthalimides were identified as tyrosine kinases inhibitors (TKIs) besides their conventional cytotoxicity.A novel series of naphthalimides were synthesized. Several of them could inhibit both topo II and angiogenesis-related receptor tyrosine kinases. The representative compound 8d not only was potent antiproliferative agent, but also exhibited effective antiangiogenic activity at 10 μM
Co-reporter:Xu Zhang, Ting Peng, Xun Ji, Jian Li, Linjiang Tong, Zeng Li, Wei Yang, Yungen Xu, Mengyuan Li, Jian Ding, Hualiang Jiang, Hua Xie, Hong Liu
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 24) pp:7988-7998
Publication Date(Web):15 December 2013
DOI:10.1016/j.bmc.2013.09.049
A novel series of anilinoquinazoline compounds with C-6 urea-linked side chains was designed and synthesized as reversible inhibitors of epidermal growth factor receptor (EGFR) based on the structure–activity relationships (SARs) of anilinoquinazoline inhibitors. All compounds demonstrated good inhibition of EGFR wild type (EGFR wt) (IC50 = 0.024–1.715 μM) and inhibited proliferation of A431cell line (IC50 = 0.116–22.008 μM). The binding mode of compounds 8a, 8d, 8k and 8o was consistent with the biological results. Moreover, compounds 8k and 8l almost completely blocked the phosphorylation of EGFR in A431 cell line at 0.01 μM. Interestingly, all of the compounds also demonstrated moderate inhibition of EGFR/T790M/L858R (IC50 = 0.049–5.578 μM). In addition, compounds 8f and 8h blocked the autophosphorylation of EGFR in NCI-H1975 cells at high concentration (10 μM), and compound 8f was confirmed to be an irreversible inhibitor through the dilution method. Importantly, the compounds with C-6 urea-linked side chains which did not contain Michael acceptors demonstrated moderate to strong irreversible EGFR inhibition.

![7-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-3,4-dihydro-2H-benzo[b][1,4]oxazine](http://img.cochemist.com/ccimg/1361200/1361110-64-6.png)
![7-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-3,4-dihydro-2H-benzo[b][1,4]oxazine](http://img.cochemist.com/ccimg/1361200/1361110-64-6_b.png)
![7-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-2H-benzo[b][1,4]oxazin-3(4H)-one](http://img.cochemist.com/ccimg/1219200/1219130-57-0.png)
![7-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-2H-benzo[b][1,4]oxazin-3(4H)-one](http://img.cochemist.com/ccimg/1219200/1219130-57-0_b.png)
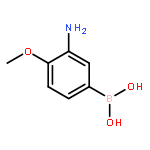
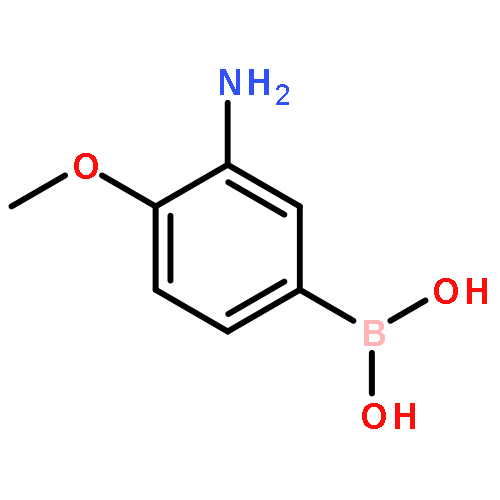
![N-(4-Chlorophenyl)-6-[(6,7-dimethoxy-4-quinolinyl)oxy]-1-naphtham ide](http://img.cochemist.com/ccimg/861900/861874-08-0.png)
![N-(4-Chlorophenyl)-6-[(6,7-dimethoxy-4-quinolinyl)oxy]-1-naphtham ide](http://img.cochemist.com/ccimg/861900/861874-08-0_b.png)
![5-Bromo-4-chlorothieno[2,3-d]pyrimidine](http://img.cochemist.com/ccimg/815000/814918-95-1.png)
![5-Bromo-4-chlorothieno[2,3-d]pyrimidine](http://img.cochemist.com/ccimg/815000/814918-95-1_b.png)
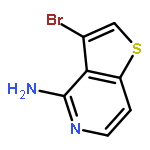
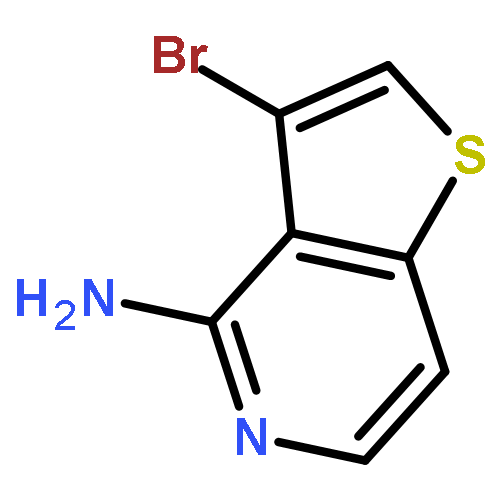


![Boronic acid, [3,5-dimethoxy-4-(phenylmethoxy)phenyl]-](http://img.cochemist.com/ccimg/482700/482627-95-2.png)
![Boronic acid, [3,5-dimethoxy-4-(phenylmethoxy)phenyl]-](http://img.cochemist.com/ccimg/482700/482627-95-2_b.png)
![Benzenamine, 3-[(methylsulfonyl)methyl]-](http://img.cochemist.com/ccimg/262000/261925-02-4.png)
![Benzenamine, 3-[(methylsulfonyl)methyl]-](http://img.cochemist.com/ccimg/262000/261925-02-4_b.png)
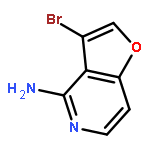
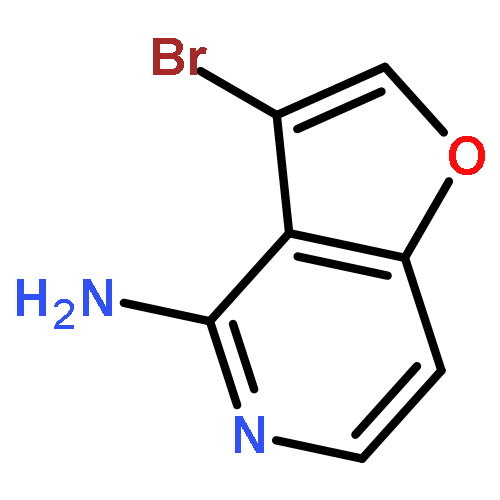
![3-Bromo-4-chlorofuro[3,2-c]pyridine](http://img.cochemist.com/ccimg/221000/220939-72-0.png)
![3-Bromo-4-chlorofuro[3,2-c]pyridine](http://img.cochemist.com/ccimg/221000/220939-72-0_b.png)
![7-Oxa-2-azaspiro[3.5]nonane](http://img.cochemist.com/ccimg/194200/194157-10-3.png)
![7-Oxa-2-azaspiro[3.5]nonane](http://img.cochemist.com/ccimg/194200/194157-10-3_b.png)


















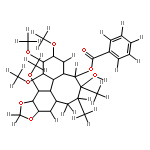
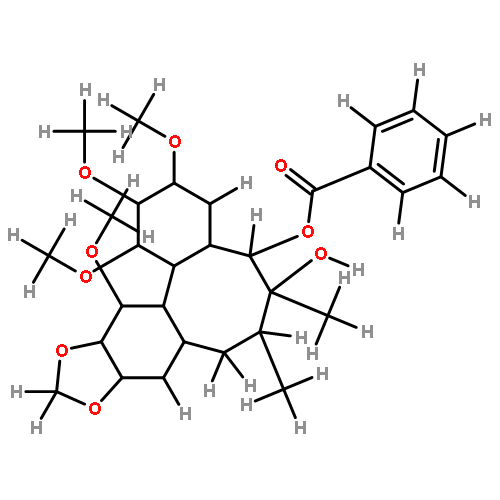


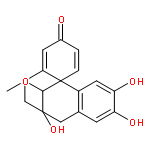
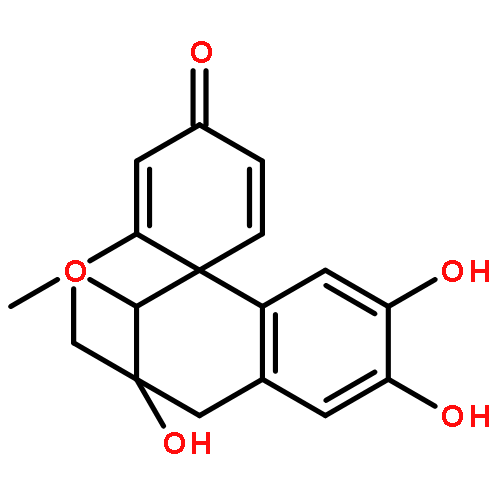


![7-Hydroxy-2H-benzo[b][1,4]oxazin-3(4H)-one](http://img.cochemist.com/ccimg/67200/67193-97-9.png)
![7-Hydroxy-2H-benzo[b][1,4]oxazin-3(4H)-one](http://img.cochemist.com/ccimg/67200/67193-97-9_b.png)


![6-NITRO-3H-THIENO[2,3-D]PYRIMIDIN-4-ONE](http://img.cochemist.com/ccimg/56900/56844-41-8.png)
![6-NITRO-3H-THIENO[2,3-D]PYRIMIDIN-4-ONE](http://img.cochemist.com/ccimg/56900/56844-41-8_b.png)
![Thieno[2,3-d]pyrimidine, 4-chloro-6-nitro-](http://img.cochemist.com/ccimg/56900/56844-13-4.png)
![Thieno[2,3-d]pyrimidine, 4-chloro-6-nitro-](http://img.cochemist.com/ccimg/56900/56844-13-4_b.png)


![(2r,3r)-2-(3,4-dihydroxyphenyl)-5,7-dihydroxy-3-[(2s,3r,4r,5r,6s)-3,4,5-trihydroxy-6-methyloxan-2-yl]oxy-2,3-dihydrochromen-4-one](http://img.cochemist.com/ccimg/29900/29838-67-3.png)
![(2r,3r)-2-(3,4-dihydroxyphenyl)-5,7-dihydroxy-3-[(2s,3r,4r,5r,6s)-3,4,5-trihydroxy-6-methyloxan-2-yl]oxy-2,3-dihydrochromen-4-one](http://img.cochemist.com/ccimg/29900/29838-67-3_b.png)
![3-Furanmethanol,tetrahydro-2-(4-hydroxy-3-methoxyphenyl)-4-[(4-hydroxy-3-methoxyphenyl)methyl]-,(2S,3R,4R)-](http://img.cochemist.com/ccimg/27100/27003-73-2.png)
![3-Furanmethanol,tetrahydro-2-(4-hydroxy-3-methoxyphenyl)-4-[(4-hydroxy-3-methoxyphenyl)methyl]-,(2S,3R,4R)-](http://img.cochemist.com/ccimg/27100/27003-73-2_b.png)
![{4-[(Methylsulfonyl)methyl]phenyl}amine hydrochloride](http://img.cochemist.com/ccimg/24200/24176-70-3.png)
![{4-[(Methylsulfonyl)methyl]phenyl}amine hydrochloride](http://img.cochemist.com/ccimg/24200/24176-70-3_b.png)


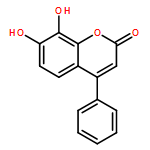
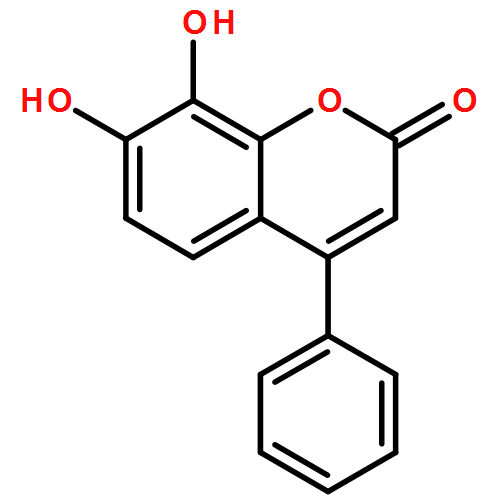
![Phenol,4,4'-(tetrahydro-1H,3H-furo[3,4-c]furan-1,4-diyl)bis[2,6-dimethoxy-](http://img.cochemist.com/ccimg/500/487-35-4.png)
![Phenol,4,4'-(tetrahydro-1H,3H-furo[3,4-c]furan-1,4-diyl)bis[2,6-dimethoxy-](http://img.cochemist.com/ccimg/500/487-35-4_b.png)


![2-Oxa-6-azaspiro[3.3]heptane](http://img.cochemist.com/ccimg/200/174-78-7.png)
![2-Oxa-6-azaspiro[3.3]heptane](http://img.cochemist.com/ccimg/200/174-78-7_b.png)
![1-Naphthalenecarboxylic acid, 6-[[(trifluoromethyl)sulfonyl]oxy]-, methylester](http://img.cochemist.com/ccimg/255100/255050-65-8.png)
![1-Naphthalenecarboxylic acid, 6-[[(trifluoromethyl)sulfonyl]oxy]-, methylester](http://img.cochemist.com/ccimg/255100/255050-65-8_b.png)
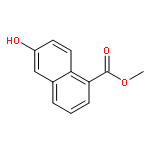
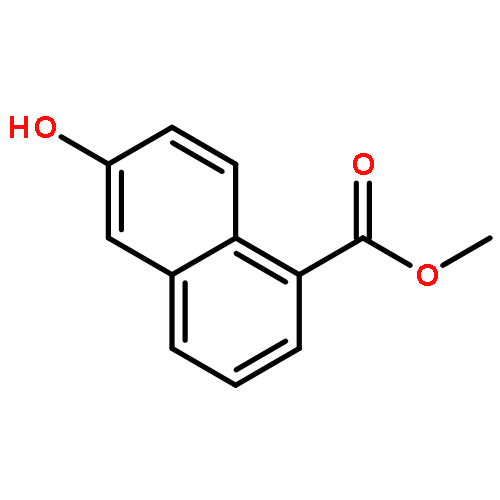
![1-[2-HYDROXY-3,4-BIS(PHENYLMETHOXY)PHENYL]ETHANONE](http://img.cochemist.com/ccimg/2700/2652-27-9.png)
![1-[2-HYDROXY-3,4-BIS(PHENYLMETHOXY)PHENYL]ETHANONE](http://img.cochemist.com/ccimg/2700/2652-27-9_b.png)