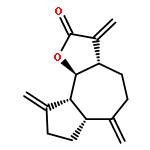
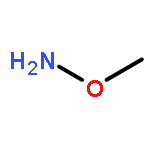
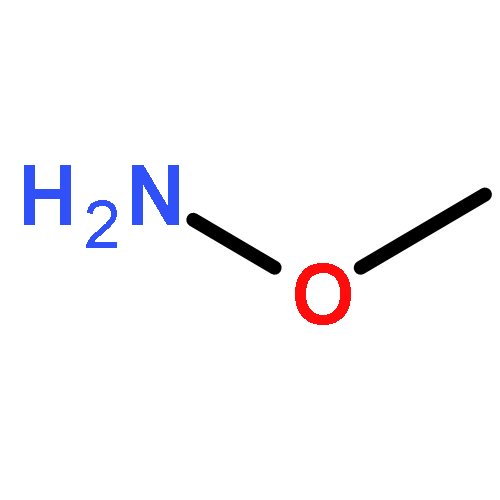
Co-reporter:Zhongjin Yang, Beijia Kuang, Ning Kang, Yahui Ding, Weizhi Ge, Lihui Lian, Yuan Gao, Yuqing Wei, Yue Chen, Quan Zhang
European Journal of Medicinal Chemistry 2017 Volume 127(Volume 127) pp:
Publication Date(Web):15 February 2017
DOI:10.1016/j.ejmech.2016.12.044
•Compound 6j is 8.7 folds more potent than that of PTL for KG1a cells.•The selectivity index of 6j between KG1a and normal cells was 30.8.•Preliminary study revealed that compound 6j could induce apoptosis of KG1a cells.Parthenolide (PTL) selectively ablates leukemia stem cells (LSCs). A series of PTL derivatives with modifications on C-14 of PTL was synthesized, and most of the derivatives showed high activities against HL-60 and KG1a. The most potent compound 6j exhibited IC50 values of 0.4 μM and 1.1 μM against KG1a and HL-60, respectively, which were 8.7 and 3.8 folds more potent than those of PTL, respectively. Moreover, compound 6j showed relatively low toxicity to normal cells (IC50 = 12.3 μM) comparing with its high anti-AML activity. The selectivity indexes for AML cells KG1a and HL-60 were 30.8 and 11.2, respectively. Preliminary study revealed that compound 6j could induce apoptosis of KG1a cells.Download high-res image (178KB)Download full-size image
Co-reporter:Yuanjun Sun;Yahui Ding;Dongmei Li;Ruifei Zhou;Xiuwen Su;Juan Yang;Xiaoqian Guo;Chuanke Chong;Jinghan Wang;Weicheng Zhang;Cuigai Bai;Liang Wang; Dr. Yue Chen
Angewandte Chemie International Edition 2017 Volume 56(Issue 46) pp:14627-14631
Publication Date(Web):2017/11/13
DOI:10.1002/anie.201709744
AbstractAsymmetric total synthesis of the cyclic depsipeptide BE-43547A2 was achieved in 15 linear steps on a 350 mg scale in one batch. The synthesis features the highly diastereoselective construction of an α-hydroxy-β-ketoamide through α-hydroxylation with a d.r. of up to 86:1. BE-43547A2 significantly reduces the percentage of pancreatic cancer stem cells (PCSCs) in Panc-1 cell cultures, and dramatically reduces the tumorsphere-forming capability of Panc-1 cells. An in vivo tumor-initiation assay, a gold standard for cancer stem cell assays, confirmed that BE-43547A2 can abolish the tumorigenesis of Panc-1 cells. The anti-PCSC activity of BE-43547A2 could make this depsipeptide scaffold a promising starting point for discovering new PCSC-targeting drugs.
Co-reporter:Jinghan Wang, Beijia Kuang, Xiaoqian Guo, Jianwei Liu, Yahui Ding, Jiangnan Li, Shende Jiang, Ying Liu, Fang Bai, Luyuan Li, Quan Zhang, Xiao-Yu Zhu, Bo Xia, Chun-Qi Li, Liang Wang, Guang Yang, and Yue Chen
Journal of Medicinal Chemistry 2017 Volume 60(Issue 3) pp:
Publication Date(Web):January 11, 2017
DOI:10.1021/acs.jmedchem.6b01745
Natural depsipeptide vinylamycin was reported to be an antibiotic previously. Herein we report vinylamycin to be active against K562 leukemia cells (IC50 = 4.86 μM) and be unstable in plasma (t1/2 = 0.54 h). A total of 24 vinylamycin analogues with modification of the OH group and chiral centers were generated via a combinatorial approach. The lead compound 1a was subsequently characterized as having the following: no antimicrobial activity, significantly higher plasma stability (t1/2 = 14.3 h), improved activity against K562 leukemia cells (IC50 = 0.64 μM), and up to 75% cell inhibition without significant toxicities in K562 cells xenograft zebrafish model. Furthermore, compound 1a maintained its activity against the breast cancer cell line MCF-7 under hypoxic conditions. In comparison, the activity of gemcitabine in the same hypoxic in vitro model of MCF-7 cells was 15-fold lower. Therefore, the present results demonstrate that 1a has great potential as an anticancer agent.
Co-reporter:Zhantao Yang, Xiaolong Xu, Chun-Hua Yang, Yunfeng Tian, Xinyi Chen, Lihui Lian, Wenwei Pan, Xuncheng Su, Weicheng Zhang, and Yue Chen
Organic Letters 2016 Volume 18(Issue 21) pp:5768-5770
Publication Date(Web):October 13, 2016
DOI:10.1021/acs.orglett.6b02729
Nannocystin A is a 21-membered cyclodepsipeptide showing remarkable anticancer properties. Described is the total synthesis of nannocystin A, which features an asymmetric vinylogous Mukaiyama aldol reaction for efficient assembly of the penultimate open-chain precursor and a pivotal intramolecular Heck cross-coupling for the final macrocyclization.
Co-reporter:Feng Sang; Yahui Ding; Jinghan Wang; Bingxia Sun; Jianlei Sun; Yan Geng; Zhang Zhang; Ke Ding; Ling-Ling Wu; Jian-Wei Liu; Cuigai Bai; Guang Yang; Quan Zhang; Lu-Yuan Li
Journal of Medicinal Chemistry 2016 Volume 59(Issue 3) pp:1184-1196
Publication Date(Web):January 27, 2016
DOI:10.1021/acs.jmedchem.5b01841
Natural product rakicidin A induces cell death in TKI-resistant chronic myelogenous leukemia (CML) cells. Therefore, 14 rakicidin A analogues were synthesized via a highly efficient combinatorial strategy and were evaluated against CML cell lines. The conjugated diene moiety was found to be crucial for the anti-CML activity of rakicidin A, and the changes in the configuration(s) at C-2, C-3, C-14, C-15, and C-16 resulted in lower levels of anti-CML activity. The most promising compound was 4-methylester rakicidin A (1a). Compared with rakicidin A, 1a exhibited 2.8-fold greater potency against the imatinib-resistant cell line K562/G+ and approximately 100-fold enhanced potency compared with that of imatinib. Furthermore, compound 1a demonstrated a significantly lower resistance index against Ba/F3 cells expressing BCR-ABLT315I than bosutinib, dasatinib, nilotinib, and ponatinib, while 1a exhibited less effect on normal hematopoietic cells. Preliminary results indicated that 1a down-regulated caspase-3 and PARP, which contributes to its K562 cell inhibitory activity.
Co-reporter:Zhantao Yang, Meiyan Ma, Chun-Hua Yang, Yuan Gao, Quan Zhang, and Yue Chen
Journal of Natural Products 2016 Volume 79(Issue 9) pp:2408-2412
Publication Date(Web):August 31, 2016
DOI:10.1021/acs.jnatprod.5b01143
Absolute configurations of the three consecutive chiral centers in the cyclic depsipeptide microtermolide A have been tentatively assigned as 2‴R, 3‴R, and 4‴R. However, on the basis of a structural comparison with vinylamycin, another depsipeptide with a unique 4-amino-2,4-pentadienolate structure, the chiral centers could also be assigned as 2‴R, 3‴R, and 4‴S. Here, the first total synthesis of microtermolide A is reported and the configurations of the three consecutive chiral centers were confirmed to be 2‴R, 3‴R, and 4‴S. A similar approach was used to determine the analogous centers in microtermolide B as 2‴R, 3‴R, and 4‴S.
Co-reporter:Yong-Tao Li, Jing-Han Wang, Cheng-Wen Pan, Fan-Fei Meng, Xiao-Qian Chu, Ya-hui Ding, Wen-Zheng Qu, Hui-ying Li, Cheng Yang, Quan Zhang, Cui-Gai Bai, Yue Chen
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 5) pp:1419-1427
Publication Date(Web):1 March 2016
DOI:10.1016/j.bmcl.2016.01.068
Three novel series of 1,2,3-triazole and 1,3,4-oxadiazole derivatives of imatinib were prepared and evaluated in vitro for their cytostatic effects against a human chronic myeloid leukemia (K562), acute myeloid leukemia (HL60), and human leukemia stem-like cell line (KG1a). The structure–activity relationship was analyzed by determining the inhibitory rate of each imatinib analog. Benzene and piperazine rings were necessary groups in these compounds for maintaining inhibitory activities against the K562 and HL60 cell lines. Introducing a trifluoromethyl group significantly enhanced the potency of the compounds against these two cell lines. Surprisingly, some compounds showed significant inhibitory activities against KG1a cells without inhibiting common leukemia cell lines (K562 and HL60). These findings suggest that these compounds are able to inhibit leukemia stem-like cells.
Co-reporter:Ya-Hui Ding, Xue Gao, Jing Long, Bei-Jia Kuang, Yue Chen, Quan Zhang
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 4) pp:1165-1168
Publication Date(Web):15 February 2016
DOI:10.1016/j.bmcl.2016.01.046
Acute myeloid leukemia (AML) is a refractory disease, and the majority of AML patients died from relapse and multidrug resistance. More and more studies demonstrate that AML stem cells play key role in multidrug resistance of AML. Here, we report a derivative of dehydrocostus lactone, that is, dispirocyclopropyldehydrocostus lactone (DDL), showed preferable cytotoxicity against a series of leukemia cell lines and AML stem cells from clinical samples of AML patient. Meanwhile, DDL demonstrated no significant toxicity to normal hematopoietic cells. Therefore, the prodrug of DDL, DMADDL, was evaluated for its in vivo anti-AML activity. The result revealed that DMADDL could inhibit the tumor growth in SCID mice tumorigenicity assay. Further study suggested that DDL induced apoptosis mainly through the up-regulation of apoptosis related protein Bax, followed by the cleavage of caspase-3, caspase-9, and PARP.Acute myeloid leukemia (AML) is a refractory disease, and the majority of AML patients died from relapse and multidrug resistance. More and more studies demonstrate that AML stem cells play key role in multidrug resistance of AML. Here, we report a derivative of dehydrocostus lactone, that is, dispirocyclopropyldehydrocostus lactone (DDL), showed preferable cytotoxicity against a series of leukemia cell lines and AML stem cells from clinical samples of AML patient. Meanwhile, DDL demonstrated no significant toxicity to normal hematopoietic cells. Therefore, the prodrug of DDL, DMADDL, was evaluated for its in vivo anti-AML activity. The result revealed that DMADDL could inhibit the tumor growth in SCID mice tumorigenicity assay. Further study suggested that DDL induced apoptosis mainly through the up-regulation of apoptosis related protein Bax, followed by the cleavage of caspase-3, caspase-9, and PARP.
Co-reporter:Jing Long, Ya-Hui Ding, Pan-Pan Wang, Quan Zhang, Yue Chen
Tetrahedron Letters 2016 Volume 57(Issue 8) pp:874-877
Publication Date(Web):24 February 2016
DOI:10.1016/j.tetlet.2016.01.039
Two new parthenolide analogues were obtained by total synthesis and assayed for their in vitro anticancer activities to study their structure–activity relationship. Based on the structure and anticancer activity results, two new SAR can be drawn: (1) replacement of the lactone moiety with lactam moiety greatly decreased the anticancer activity; (2) the C-14 methyl group of parthenolide might be important for its high anticancer activity.Two new parthenolide analogues were obtained by total synthesis and assayed for their in vitro anticancer activities to study their structure–activity relationship. Based on the structure and anticancer activity results, two new SAR can be drawn: (1) replacement of the lactone moiety with lactam moiety greatly decreased the anticancer activity; (2) the C-14 methyl group of parthenolide might be important for its high anticancer activity.
Co-reporter:Zhantao Yang, Guang Yang, Meiyan Ma, Jiangnan Li, Jianwei Liu, Jinghan Wang, Shende Jiang, Quan Zhang, and Yue Chen
Organic Letters 2015 Volume 17(Issue 23) pp:5725-5727
Publication Date(Web):November 2, 2015
DOI:10.1021/acs.orglett.5b02809
The absolute configurations of the three unknown chiral centers in vinylamycin were predicted according to the structural comparison with microtermolide A and rakicidin A, and then total syntheses of vinylamycin were applied to determine the three unknown chiral centers as 14R, 15R, and 16S.
Co-reporter:Zhong-Jin Yang; Wei-Zhi Ge; Qiu-Ying Li; Yaxin Lu; Jian-Miao Gong; Bei-Jia Kuang; Xiaonan Xi; Haiting Wu; Quan Zhang
Journal of Medicinal Chemistry 2015 Volume 58(Issue 17) pp:7007-7020
Publication Date(Web):July 30, 2015
DOI:10.1021/acs.jmedchem.5b00915
Inspired by the biosynthesis of sesquiterpene lactones (SLs), herein we report the asymmetric total synthesis of the germacrane ring (24). The synthetic strategy features a selective aldol reaction between β,γ-unsaturated chiral sulfonylamide 15a and aldehyde 13, as well as the intramolecular α-alkylation of sulfone 21 to construct a 10-membered carbocylic ring. The key intermediate 24 can be used to prepare the natural products costunolide and parthenolide (PTL), which are the key precursors for transformation into other SLs. Furthermore, the described synthetic sequences are amenable to the total synthesis of SL analogues, such as trifluoromethylated analogues 32 and 45. Analogues 32 and 45 maintained high activities against a series of cancer cell lines compared to their parent PTL and costunolide, respectively. In addition, 32 showed enhanced tolerance to acidic media compared with PTL. To our surprise, PTL and 32 showed comparable half-lives in rat plasma and in the presence of human liver microsomes.
Co-reporter:Feng Sang ; Dongmei Li ; Xiaolong Sun ; Xianqiang Cao ; Liang Wang ; Jianlei Sun ; Bingxia Sun ; Lingling Wu ; Guang Yang ; Xiaoqian Chu ; Jinghan Wang ; Changming Dong ; Yan Geng ; Hong Jiang ; Haibo Long ; Sijia Chen ; Guiyan Wang ; Shuzhong Zhang ; Quan Zhang
Journal of the American Chemical Society 2014 Volume 136(Issue 44) pp:15787-15791
Publication Date(Web):October 6, 2014
DOI:10.1021/ja509379j
Rakicidin A is a cyclic depsipeptide that has exhibited unique growth inhibitory activity against chronic myelogenous leukemia stem cells. Furthermore, rakicidin A has five chiral centers with unknown stereochemical assignment, and thus, can be represented by one of 32 possible stereoisomers. To predict the most probable stereochemistry of rakicidin A, calculations and structural comparison with natural cyclic depsipeptides were applied. A total synthesis of the proposed structure was subsequently completed and highlighted by the creation of a sterically hindered ester bond (C1–C15) through trans-acylation from an easily established isomer (C1–C13). The analytic data of the synthetic target were consistent with that of natural rakicidin A, and then the absolute configuration of rakicidin A was assigned as 2S, 3S, 14S, 15S, 16R. This work suggests strategies for the determination of unknown chiral centers in other cyclic depsipeptides, such as rakicidin B, C, D, BE-43547, and vinylamycin, and facilitates the investigations of rakicidin A as an anticancer stem cell agent.
Co-reporter:Jing Long ; Shan-Feng Zhang ; Pan-Pan Wang ; Xue-Mei Zhang ; Zhong-Jin Yang ; Quan Zhang
Journal of Medicinal Chemistry 2014 Volume 57(Issue 16) pp:7098-7112
Publication Date(Web):August 7, 2014
DOI:10.1021/jm5009456
The first total synthesis of parthenolide (1) is described. The key feature of this synthesis is the formation of a 10-membered carbocylic ring by a macrocyclic stereocontrolled Barbier reaction, followed by a photoinduced Z/E isomerization. The biological evaluation of a small library of parthenolide analogues (19, 33, and 34) disclosed a preliminary structure–activity relationship (SAR). The results revealed that the C1, C10 double bond configuration of parthenolide has little or no effect on the activity, and the C6 and C7 configurations of the lactone ring have a moderate impact on the activities against some cancer cell lines.
Co-reporter:Ningning Li, Yintong Xu, Qiang Xia, Cuigai Bai, Taiyi Wang, Lei Wang, Dingdi He, Nannan Xie, Lixin Li, Jing Wang, Hong-Gang Zhou, Feng Xu, Cheng Yang, Quan Zhang, Zheng Yin, Yu Guo, Yue Chen
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 1) pp:386-389
Publication Date(Web):1 January 2014
DOI:10.1016/j.bmcl.2013.10.068
Captopril is a New Delhi metallo-β-lactamase-1 (NDM-1) inhibitor with an IC50 value of 7.9 μM. It is composed of two units: a 3-mercapto-2-methylpropanoyl fragment and a proline residue. In this study, we synthesized simple amide derivatives of 3-mercapto-2-methylpropanoic acid, and then tested them as NDM-1 inhibitors in order to identify the pharmacophore for NDM-1 inhibition. We found that the lead compound 22 had an IC50 value of 1.0 μM. Further structure simplification provided compounds 31 and 32, which had IC50 values of 15 and 10 μM, respectively. As compound 32 is a clinically used antidote for metal poisoning, it has great potential to be repurposed to treat bacterial infections.Captopril is a New Delhi metallo-β-lactamase-1 (NDM-1) inhibitor with an IC50 value of 7.9 μM. It is composed of two units: a 3-mercapto-2-methylpropanoyl fragment and a proline residue. In this study, we synthesized simple amide derivatives of 3-mercapto-2-methylpropanoic acid, and then tested them as NDM-1 inhibitors in order to identify the pharmacophore for NDM-1 inhibition. We found that the lead compound 22 had an IC50 value of 1.0 μM. Further structure simplification provided compounds 31 and 32, which had IC50 values of 15 and 10 μM, respectively. As compound 32 is a clinically used antidote for metal poisoning, it has great potential to be repurposed to treat bacterial infections.
Co-reporter:Feng Sang, Peng Feng, Jie Chen, Yahui Ding, Xiyan Duan, Jiadai Zhai, Xiaoyan Ma, Bin Zhang, Quan Zhang, Jianping Lin, Yue Chen
European Journal of Medicinal Chemistry 2013 Volume 68() pp:321-332
Publication Date(Web):October 2013
DOI:10.1016/j.ejmech.2013.08.003
•A highly efficient approach was applied to synthesize Epo D and its new analogues.•The new analogues demonstrated significant difference in biological activity.•The conformations of Epo D and its new analogues were investigated.Epothilone D (Epo D) and its 9-Methyl conformational analogues were synthesized through a highly efficient combinatorial approach. The fragment E was synthesized in 11 total steps with 6 longest linear steps, and each aldehyde B was prepared via a 3-step sequence. Starting from the common precursor E and a suitable aldehydes B, each target molecule were obtained in only 4 steps. The 9-(S)-epo D and 9-(R)-epo D demonstrated significant difference in inhibition activities against cancer cell lines and in conformational analysis.A highly efficient combinatorial approach was applied to synthesize epothilone D and its 9-Methyl analogues.
Co-reporter:Jing-Han Wang, Cheng-Wen Pan, Yong-Tao Li, Fan-Fei Meng, Hong-Gang Zhou, Cheng Yang, Quan Zhang, Cui-Gai Bai, Yue Chen
Tetrahedron Letters 2013 Volume 54(Issue 26) pp:3406-3409
Publication Date(Web):26 June 2013
DOI:10.1016/j.tetlet.2013.04.067
Triazole moiety is frequently employed in drug discovery and optimization. However, most syntheses of triazole drugs involve isolation of highly explosive azides. Herein we report safe and high efficient syntheses of triazole drugs in aqueous/organic solvent systems with Cu2O nanoparticle (Cu2O-NP) as the catalyst of azide–alkyne cycloaddition (CuAAC). Since Cu2O-NP can be efficiently dispensed in aqueous and some organic solvents, the azide solutions from the previous preparation could be used directly in the next CuAAC stage without isolation. Therefore, this synthetic strategy is safe, convenient, and high yielding for the syntheses of triazole drugs.
Co-reporter:Ya-Hui Ding, Hong-Xia Fan, Jing Long, Quan Zhang, Yue Chen
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 22) pp:6087-6092
Publication Date(Web):15 November 2013
DOI:10.1016/j.bmcl.2013.09.028
A series of guaianolide-type sesquiterpene lactones derivatives with arylation of α-methylene-γ-lactone moiety was synthesized using Heck reactions, and was evaluated for their activities against acute myelogenous leukemia (AML) cell line HL-60 and doxorubicin-resistant cell line HL-60/A. Although all compounds were significantly less active against HL-60 than the parent molecules, surprisingly, compounds 3a, 4c–4e, 5e, and 8d exhibited high potency against doxorubicin-resistant cell line HL-60/A (IC50 = 6.2–19 μM), and their activities against HL-60/A were comparable to that of their parent molecules. In view of their novel activities against HL-60/A, compound 5e with inhibitory activity against HL-60/A (IC50 = 6.2 ± 0.5 μM) was selected for study its preliminary mechanism. The result reveals that compound 5e can obviously induce apoptosis.A series of guaianolide-type sesquiterpene lactones derivatives with arylation of α-methylene-γ-lactone moiety was synthesized using Heck reactions, and was evaluated for their activities against acute myelogenous leukemia (AML) cell line HL-60 and doxorubicin-resistant cell line HL-60/A. Although all compounds were significantly less active against HL-60 than the parent molecules, surprisingly, compounds 3a, 4c–4e, 5e, and 8d exhibited high potency against doxorubicin-resistant cell line HL-60/A (IC50 = 6.2–19 μM), and their activities against HL-60/A were comparable to that of their parent molecules. In view of their novel activities against HL-60/A, compound 5e with inhibitory activity against HL-60/A (IC50 = 6.2 ± 0.5 μM) was selected for study its preliminary mechanism. The result reveals that compound 5e can obviously induce apoptosis.
Co-reporter:Xiudong Yang, Changming Dong, Jian Chen, Qi Liu, Bin Han, Quan Zhang, Yue Chen
Tetrahedron Letters 2013 Volume 54(Issue 23) pp:2986-2988
Publication Date(Web):5 June 2013
DOI:10.1016/j.tetlet.2013.03.127
A tubulysin V analogue with triazole/Me modified Tuv fragment was synthesized in 14 linear steps. The triazoltubulysin analogue maintains significant biological activities against several cancer cell lines, and then provides a new insight into the biological conformation of tubulysins.
Co-reporter:Jing Long, Ya-Hui Ding, Pan-Pan Wang, Quan Zhang, and Yue Chen
The Journal of Organic Chemistry 2013 Volume 78(Issue 20) pp:10512-10518
Publication Date(Web):September 18, 2013
DOI:10.1021/jo401606q
Parthenolide showed extensive bioactivities including selective eradication of AML stem cells. Herein we report protection-free semisyntheses of parthenolide and its cyclopropyl analogue (compound 10) from the abundant natural product costunolide with an overall yield of 55 and 60%, respectively. Compound 10 was more stable than parthenolide, and it maintained comparable activities against AML cell lines and AML stem cells. Therefore, compound 10 might be a superior small molecule than parthenolide as a tool for investigation of cancer stem cell biology.
Co-reporter:Xiu-dong Yang;Chang-ming Dong;Jian Chen;Ya-hui Ding;Qi Liu;Xiao-yan Ma;Dr. Quan Zhang; Yue Chen
Chemistry – An Asian Journal 2013 Volume 8( Issue 6) pp:1213-1222
Publication Date(Web):
DOI:10.1002/asia.201300051
Abstract
The Tup fragments of tubulysins were synthesized with a tandem reaction as the key step, and unexpected diastereoselectivity was observed in the first Grignard addition stage. The coupling of the enolate of a thiazolyl ketone with chiral sulfinimines furnished the backbone of the Tuv fragment with over 100:1 d.r. and high yield. Thus, tubulysin U and C-4 epi-tubulysin U were prepared in a highly selective and efficient manner. The results of the MTT assay furthermore indicated that C-4 epi-tubulysin U maintained significant growth inhibition activities against several cancer cell lines.
Co-reporter:Quan Zhang ; Yaxin Lu ; Yahui Ding ; Jiadai Zhai ; Qing Ji ; Weiwei Ma ; Ming Yang ; Hongxia Fan ; Jing Long ; Zhongsheng Tong ; Yehui Shi ; Yongsheng Jia ; Bin Han ; Wenpeng Zhang ; Chuanjiang Qiu ; Xiaoyan Ma ; Qiuying Li ; Qianqian Shi ; Haoliang Zhang ; Dongmei Li ; Jing Zhang ; Jianping Lin ; Lu-Yuan Li ; Yingdai Gao
Journal of Medicinal Chemistry 2012 Volume 55(Issue 20) pp:8757-8769
Publication Date(Web):September 17, 2012
DOI:10.1021/jm301064b
Small molecules that can selectively target cancer stem cells (CSCs) remain rare currently and exhibit no common structural features. Here we report a series of guaianolide sesquiterpene lactones (GSLs) and their derivatives that can selectively eradicate acute myelogenous leukemia (AML) stem or progenitor cells. Natural GSL compounds arglabin, an anticancer clinical drug, and micheliolide (MCL), are able to reduce the proportion of AML stem cells (CD34+CD38–) in primary AML cells. Targeting of AML stem cells is further confirmed by a sharp reduction of colony-forming units of primary AML cells upon MCL treatment. Moreover, DMAMCL, the dimethylamino Michael adduct of MCL, slowly releases MCL in plasma and in vivo and demonstrates remarkable therapeutic efficacy in the nonobese diabetic/severe combined immunodeficiency AML models. These findings indicate that GSL is an ample source for chemical agents against AML stem or progenitor cells and that GSL is potentially highly useful to explore anti-CSC approaches.
Co-reporter:Xiyan Duan;Yan Zhang;Yahui Ding;Jianping Lin;Xianglei Kong;Quan Zhang;Changming Dong;Guoan Luo
European Journal of Organic Chemistry 2012 Volume 2012( Issue 3) pp:500-508
Publication Date(Web):
DOI:10.1002/ejoc.201101306
Abstract
We report the total synthesis of a triazole-epothilone analogue 1. The key step to generate the macrocyclic ring and the triazole ring was to apply Cu2O nanoparticles (Cu2O-NPs) to catalyze the 1,3-dipolar cycloaddition. The conformation of 1 and its bioactivity in MCF cancer cell lines were investigated.
Co-reporter:Jia-Dai Zhai, Dongmei Li, Jing Long, Hao-Liang Zhang, Jian-Ping Lin, Chuan-Jiang Qiu, Quan Zhang, and Yue Chen
The Journal of Organic Chemistry 2012 Volume 77(Issue 16) pp:7103-7107
Publication Date(Web):July 31, 2012
DOI:10.1021/jo300888s
The semisynthesis of arglabin, an anticancer drug in clinical application, is developed from abundant natural product parthenolide via three steps. Each step in this sequence is highly stereoselective, and the substrate-dependent stereoselectivity in the epoxidation step can be explained by computational calculations. The success of chemical semisynthesis of arglabin suggests that the biosynthesis of arglabin might proceed in a similar pathway.
Co-reporter:Zhenfang Zhang;Changming Dong;Cuihong Yang;Di Hu;Jing Long;Ling Wang;He Li;Deling Kong
Advanced Synthesis & Catalysis 2010 Volume 352( Issue 10) pp:1600-1604
Publication Date(Web):
DOI:10.1002/adsc.201000206
Abstract
A novel form of polyvinylpyrrolidone (PVP) coated copper(I) oxide nanoparticle (Cu2O-NP) was prepared and used to catalyze azide-alkyne click reactions in water under aerobic conditions. The nanoparticles were well dispersed in aqueous solutions and have a size of 20±10 nm, as determined by transmission electron microscope (TEM). Inductively coupled plasma (ICP), X-ray powder diffraction (XRD), and X-ray photoelectron spectroscopy (XPS) analyses demonstrated that the main content of Cu2O-NP is copper(I). The cytotoxicity of it was evaluated by an in vitro 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay and its catalytic efficiency for azide-alkyne click reactions was studied in water and organic solvents at physiological temperatures. Our results indicate that Cu2O-NP is more efficient in catalytic reactions in water for both aliphatic and aromatic azides and alkynes and less toxic than the commonly used CuSO4/reductant catalyst systems.

![Carbamic acid, N-[(1R)-2-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-1-(hydroxymethyl)ethyl]-, 1,1-dimethylethyl ester](/data/chemimg/1781600/198417-69-5.png)
![Carbamic acid, N-[(1R)-2-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-1-(hydroxymethyl)ethyl]-, 1,1-dimethylethyl ester](/data/chemimg/1781600/198417-69-5_b.png)
![Carbamic acid, N-[(1S)-2-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-1-formylethyl]-, 1,1-dimethylethyl ester](/data/chemimg/1740100/154456-41-4.png)
![Carbamic acid, N-[(1S)-2-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-1-formylethyl]-, 1,1-dimethylethyl ester](/data/chemimg/1740100/154456-41-4_b.png)
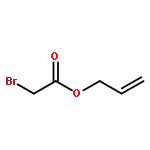
![Phosphonium, [2-oxo-2-(2-propenyloxy)ethyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/501100/501034-84-0.png)
![Phosphonium, [2-oxo-2-(2-propenyloxy)ethyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/501100/501034-84-0_b.png)


![Benzenemethanol, α-[(1R)-1-methyl-2-propen-1-yl]-, (αR)-](/data/chemimg/110800/121842-16-8.png)
![Benzenemethanol, α-[(1R)-1-methyl-2-propen-1-yl]-, (αR)-](/data/chemimg/110800/121842-16-8_b.png)




















![2-Pyrimidinamine, N-[2-methyl-5-[1-[2-(4-morpholinyl)-5-(trifluoromethyl)phenyl]-1H-1,2,3-triazol-4-yl]phenyl]-4-(3-pyridinyl)-](/data/chemimg/3721800/1877281-19-0.png)
![2-Pyrimidinamine, N-[2-methyl-5-[1-[2-(4-morpholinyl)-5-(trifluoromethyl)phenyl]-1H-1,2,3-triazol-4-yl]phenyl]-4-(3-pyridinyl)-](/data/chemimg/3721800/1877281-19-0_b.png)
![2-Pyrimidinamine, N-[2-methyl-5-[1-[2-(4-morpholinyl)phenyl]-1H-1,2,3-triazol-4-yl]phenyl]-4-(3-pyridinyl)-](/data/chemimg/3721800/1877281-18-9.png)
![2-Pyrimidinamine, N-[2-methyl-5-[1-[2-(4-morpholinyl)phenyl]-1H-1,2,3-triazol-4-yl]phenyl]-4-(3-pyridinyl)-](/data/chemimg/3721800/1877281-18-9_b.png)
![Morpholine, 4-[2-azido-4-(trifluoromethyl)phenyl]-](/data/chemimg/3723100/1877281-17-8.png)
![Morpholine, 4-[2-azido-4-(trifluoromethyl)phenyl]-](/data/chemimg/3723100/1877281-17-8_b.png)
![Pyrrolidine, 1-[(4-azidophenyl)methyl]-](/data/chemimg/3723100/1877281-16-7.png)
![Pyrrolidine, 1-[(4-azidophenyl)methyl]-](/data/chemimg/3723100/1877281-16-7_b.png)
![Piperidine, 1-[(4-azidophenyl)methyl]-](/data/chemimg/3723300/1877281-15-6.png)
![Piperidine, 1-[(4-azidophenyl)methyl]-](/data/chemimg/3723300/1877281-15-6_b.png)
![Piperazine, 1-[[4-azido-2-(trifluoromethyl)phenyl]methyl]-4-methyl-](/data/chemimg/3723100/1877281-14-5.png)
![Piperazine, 1-[[4-azido-2-(trifluoromethyl)phenyl]methyl]-4-methyl-](/data/chemimg/3723100/1877281-14-5_b.png)
![Piperazine, 1-[(4-azidophenyl)methyl]-4-methyl-](/data/chemimg/3723100/1446139-85-0.png)
![Piperazine, 1-[(4-azidophenyl)methyl]-4-methyl-](/data/chemimg/3723100/1446139-85-0_b.png)
![2-Pyrimidinamine, N-[2-methyl-5-[1-[4-(1-pyrrolidinylmethyl)phenyl]-1H-1,2,3-triazol-4-yl]phenyl]-4-(3-pyridinyl)-](/data/chemimg/3722600/1446139-84-9.png)
![2-Pyrimidinamine, N-[2-methyl-5-[1-[4-(1-pyrrolidinylmethyl)phenyl]-1H-1,2,3-triazol-4-yl]phenyl]-4-(3-pyridinyl)-](/data/chemimg/3722600/1446139-84-9_b.png)
![2-Pyrimidinamine, N-[5-[1-[4-(diethylamino)phenyl]-1H-1,2,3-triazol-4-yl]-2-methylphenyl]-4-(3-pyridinyl)-](/data/chemimg/3722600/1446139-83-8.png)
![2-Pyrimidinamine, N-[5-[1-[4-(diethylamino)phenyl]-1H-1,2,3-triazol-4-yl]-2-methylphenyl]-4-(3-pyridinyl)-](/data/chemimg/3722600/1446139-83-8_b.png)
![2-Pyrimidinamine, N-[2-methyl-5-[1-[4-(4-morpholinyl)phenyl]-1H-1,2,3-triazol-4-yl]phenyl]-4-(3-pyridinyl)-](/data/chemimg/3722600/1446139-82-7.png)
![2-Pyrimidinamine, N-[2-methyl-5-[1-[4-(4-morpholinyl)phenyl]-1H-1,2,3-triazol-4-yl]phenyl]-4-(3-pyridinyl)-](/data/chemimg/3722600/1446139-82-7_b.png)
![2-Pyrimidinamine, N-[2-methyl-5-[1-[4-(4-morpholinylmethyl)phenyl]-1H-1,2,3-triazol-4-yl]phenyl]-4-(3-pyridinyl)-](/data/chemimg/3722600/1446139-81-6.png)
![2-Pyrimidinamine, N-[2-methyl-5-[1-[4-(4-morpholinylmethyl)phenyl]-1H-1,2,3-triazol-4-yl]phenyl]-4-(3-pyridinyl)-](/data/chemimg/3722600/1446139-81-6_b.png)
![2-Pyrimidinamine, N-[2-methyl-5-[1-[4-(1-piperidinylmethyl)phenyl]-1H-1,2,3-triazol-4-yl]phenyl]-4-(3-pyridinyl)-](/data/chemimg/3722700/1446139-80-5.png)
![2-Pyrimidinamine, N-[2-methyl-5-[1-[4-(1-piperidinylmethyl)phenyl]-1H-1,2,3-triazol-4-yl]phenyl]-4-(3-pyridinyl)-](/data/chemimg/3722700/1446139-80-5_b.png)
![2-Pyrimidinamine, N-[2-methyl-5-[1-[4-[(4-methyl-1-piperazinyl)methyl]-3-(trifluoromethyl)phenyl]-1H-1,2,3-triazol-4-yl]phenyl]-4-(3-pyridinyl)-](/data/chemimg/3722600/1446139-79-2.png)
![2-Pyrimidinamine, N-[2-methyl-5-[1-[4-[(4-methyl-1-piperazinyl)methyl]-3-(trifluoromethyl)phenyl]-1H-1,2,3-triazol-4-yl]phenyl]-4-(3-pyridinyl)-](/data/chemimg/3722600/1446139-79-2_b.png)
![2-Pyrimidinamine, N-[2-methyl-5-[1-[4-[(4-methyl-1-piperazinyl)methyl]phenyl]-1H-1,2,3-triazol-4-yl]phenyl]-4-(3-pyridinyl)-](/data/chemimg/3722700/1446139-78-1.png)
![2-Pyrimidinamine, N-[2-methyl-5-[1-[4-[(4-methyl-1-piperazinyl)methyl]phenyl]-1H-1,2,3-triazol-4-yl]phenyl]-4-(3-pyridinyl)-](/data/chemimg/3722700/1446139-78-1_b.png)
![2-Pyrimidinamine, N-[5-[4-[4-methoxy-3-(trifluoromethyl)phenyl]-1H-1,2,3-triazol-1-yl]-2-methylphenyl]-4-(3-pyridinyl)-](/data/chemimg/3722600/1445858-84-3.png)
![2-Pyrimidinamine, N-[5-[4-[4-methoxy-3-(trifluoromethyl)phenyl]-1H-1,2,3-triazol-1-yl]-2-methylphenyl]-4-(3-pyridinyl)-](/data/chemimg/3722600/1445858-84-3_b.png)
![2-Pyrimidinamine, N-[2-methyl-5-[4-[4-methyl-3-(trifluoromethyl)phenyl]-1H-1,2,3-triazol-1-yl]phenyl]-4-(3-pyridinyl)-](/data/chemimg/3722600/1445858-82-1.png)
![2-Pyrimidinamine, N-[2-methyl-5-[4-[4-methyl-3-(trifluoromethyl)phenyl]-1H-1,2,3-triazol-1-yl]phenyl]-4-(3-pyridinyl)-](/data/chemimg/3722600/1445858-82-1_b.png)
![2-Pyrimidinamine, N-[5-[4-[3,5-bis(trifluoromethyl)phenyl]-1H-1,2,3-triazol-1-yl]-2-methylphenyl]-4-(3-pyridinyl)-](/data/chemimg/3722600/1445858-80-9.png)
![2-Pyrimidinamine, N-[5-[4-[3,5-bis(trifluoromethyl)phenyl]-1H-1,2,3-triazol-1-yl]-2-methylphenyl]-4-(3-pyridinyl)-](/data/chemimg/3722600/1445858-80-9_b.png)
![1H-1,2,3-Triazole-4-butanol, 1-[4-methyl-3-[[4-(3-pyridinyl)-2-pyrimidinyl]amino]phenyl]-](/data/chemimg/3722600/1445858-76-3.png)
![1H-1,2,3-Triazole-4-butanol, 1-[4-methyl-3-[[4-(3-pyridinyl)-2-pyrimidinyl]amino]phenyl]-](/data/chemimg/3722600/1445858-76-3_b.png)
![1H-1,2,3-Triazole-4-methanol, α-methyl-1-[4-methyl-3-[[4-(3-pyridinyl)-2-pyrimidinyl]amino]phenyl]-](/data/chemimg/3722600/1445858-75-2.png)
![1H-1,2,3-Triazole-4-methanol, α-methyl-1-[4-methyl-3-[[4-(3-pyridinyl)-2-pyrimidinyl]amino]phenyl]-](/data/chemimg/3722600/1445858-75-2_b.png)
![2-Pyrimidinamine, N-[2-methyl-5-[4-[4-[(4-methyl-1-piperazinyl)methyl]-3-(trifluoromethyl)phenyl]-1H-1,2,3-triazol-1-yl]phenyl]-4-(3-pyridinyl)-](/data/chemimg/3722600/1445858-72-9.png)
![2-Pyrimidinamine, N-[2-methyl-5-[4-[4-[(4-methyl-1-piperazinyl)methyl]-3-(trifluoromethyl)phenyl]-1H-1,2,3-triazol-1-yl]phenyl]-4-(3-pyridinyl)-](/data/chemimg/3722600/1445858-72-9_b.png)
![1H-1,2,3-Triazole-4-ethanol, 1-[4-methyl-3-[[4-(3-pyridinyl)-2-pyrimidinyl]amino]phenyl]-](/data/chemimg/145400/1441640-67-0.png)
![1H-1,2,3-Triazole-4-ethanol, 1-[4-methyl-3-[[4-(3-pyridinyl)-2-pyrimidinyl]amino]phenyl]-](/data/chemimg/145400/1441640-67-0_b.png)
![Morpholine, 4-[(4-azidophenyl)methyl]-](/data/chemimg/3722900/1383706-25-9.png)
![Morpholine, 4-[(4-azidophenyl)methyl]-](/data/chemimg/3722900/1383706-25-9_b.png)
![2-Pyrimidinamine, N-[2-methyl-5-[4-[4-[(4-methyl-1-piperazinyl)methyl]phenyl]-1H-1,2,3-triazol-1-yl]phenyl]-4-(3-pyridinyl)-](/data/chemimg/145400/1346617-86-4.png)
![2-Pyrimidinamine, N-[2-methyl-5-[4-[4-[(4-methyl-1-piperazinyl)methyl]phenyl]-1H-1,2,3-triazol-1-yl]phenyl]-4-(3-pyridinyl)-](/data/chemimg/145400/1346617-86-4_b.png)
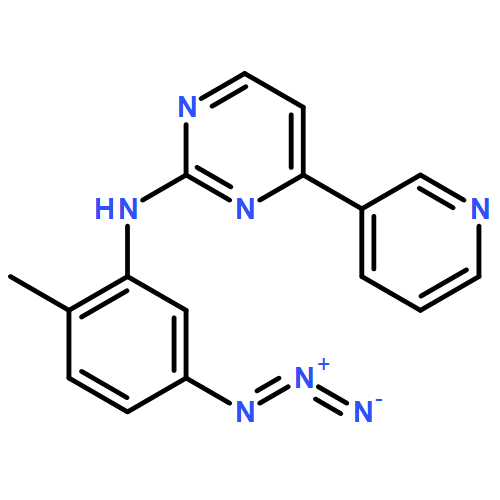




![4-[(4-methyl-1-piperazinyl)methyl]-3-(trifluoromethyl)benzoic Aci D](http://img.cochemist.com/ccimg/859300/859282-11-4.png)
![4-[(4-methyl-1-piperazinyl)methyl]-3-(trifluoromethyl)benzoic Aci D](http://img.cochemist.com/ccimg/859300/859282-11-4_b.png)


![1-Propanol, 2-amino-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (2S)-](http://img.cochemist.com/ccimg/566200/566199-97-1.png)
![1-Propanol, 2-amino-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (2S)-](http://img.cochemist.com/ccimg/566200/566199-97-1_b.png)


![Benzaldehyde,4-[(4-methyl-1-piperazinyl)methyl]-](http://img.cochemist.com/ccimg/439700/439691-80-2.png)
![Benzaldehyde,4-[(4-methyl-1-piperazinyl)methyl]-](http://img.cochemist.com/ccimg/439700/439691-80-2_b.png)
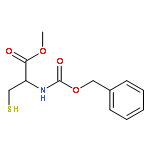
![Homocysteine, N-[(phenylmethoxy)carbonyl]-, methyl ester](http://img.cochemist.com/ccimg/382700/382601-87-8.png)
![Homocysteine, N-[(phenylmethoxy)carbonyl]-, methyl ester](http://img.cochemist.com/ccimg/382700/382601-87-8_b.png)
![4-[(4-METHOXYPHENYL)METHOXY]BUTANOIC ACID](http://img.cochemist.com/ccimg/342900/342893-41-8.png)
![4-[(4-METHOXYPHENYL)METHOXY]BUTANOIC ACID](http://img.cochemist.com/ccimg/342900/342893-41-8_b.png)






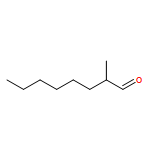
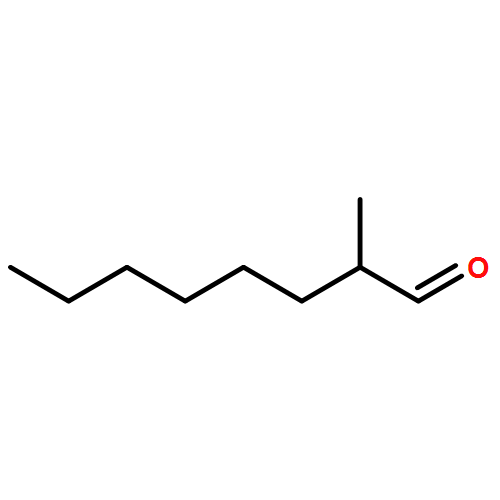
![2-Hexen-1-ol, 6-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-3-methyl-, (2E)-](http://img.cochemist.com/ccimg/187400/187336-22-7.png)
![2-Hexen-1-ol, 6-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-3-methyl-, (2E)-](http://img.cochemist.com/ccimg/187400/187336-22-7_b.png)










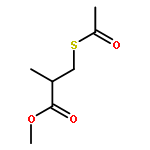

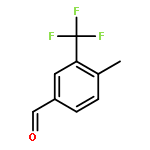





![S-[3-(benzylamino)-2-methyl-3-oxopropyl] ethanethioate](http://img.cochemist.com/ccimg/86000/85980-47-8.png)
![S-[3-(benzylamino)-2-methyl-3-oxopropyl] ethanethioate](http://img.cochemist.com/ccimg/86000/85980-47-8_b.png)
![ETHANETHIOIC ACID, S-[2-METHYL-3-OXO-3-(PHENYLAMINO)PROPYL] ESTER](http://img.cochemist.com/ccimg/86000/85980-46-7.png)
![ETHANETHIOIC ACID, S-[2-METHYL-3-OXO-3-(PHENYLAMINO)PROPYL] ESTER](http://img.cochemist.com/ccimg/86000/85980-46-7_b.png)






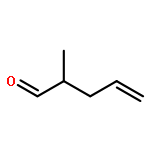



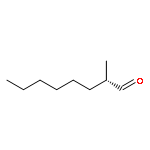
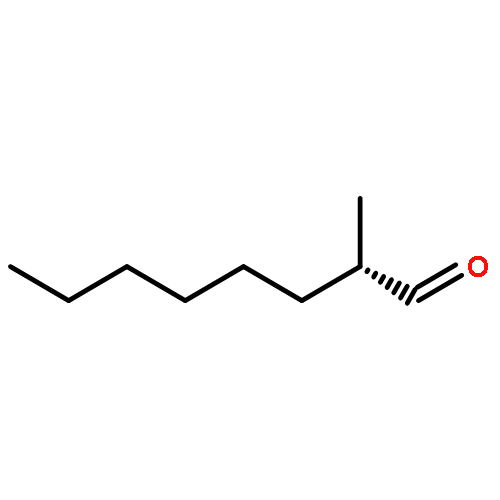
![L-Cysteine,N-[(phenylmethoxy)carbonyl]-](http://img.cochemist.com/ccimg/54000/53907-29-2.png)
![L-Cysteine,N-[(phenylmethoxy)carbonyl]-](http://img.cochemist.com/ccimg/54000/53907-29-2_b.png)










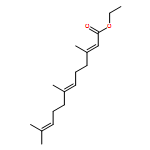
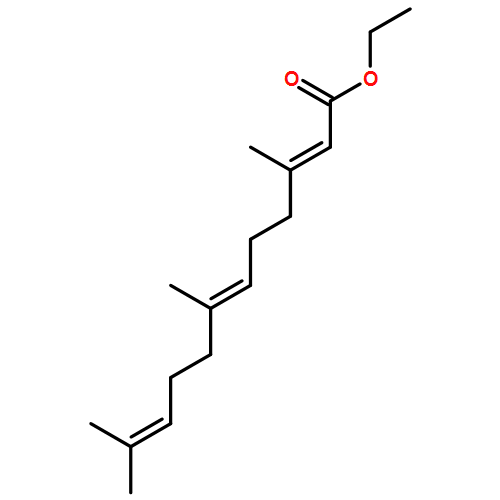
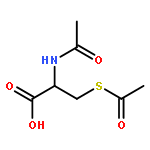

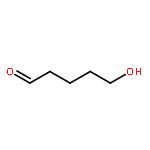





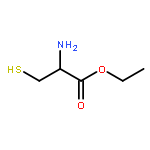
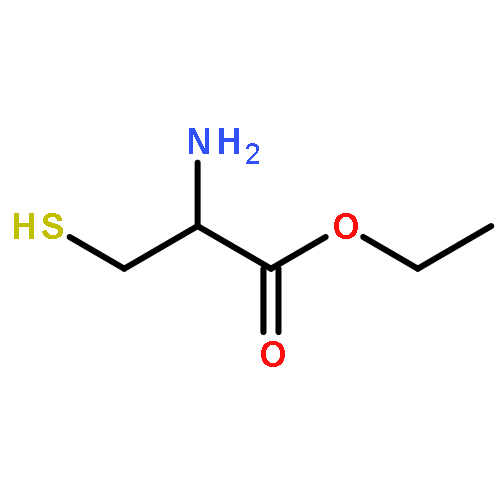
![Oxireno[9,10]cyclodeca[1,2-b]furan-9(1aH)-one,2,3,6,7,7a,8,10a,10b-octahydro-1a,5,8-trimethyl-, (1aR,4E,7aS,8S,10aS,10bR)-](http://img.cochemist.com/ccimg/2600/2513-76-0.png)
![Oxireno[9,10]cyclodeca[1,2-b]furan-9(1aH)-one,2,3,6,7,7a,8,10a,10b-octahydro-1a,5,8-trimethyl-, (1aR,4E,7aS,8S,10aS,10bR)-](http://img.cochemist.com/ccimg/2600/2513-76-0_b.png)