Co-reporter:Masashi Kaneko; Sunao Miyashita
Inorganic Chemistry 2015 Volume 54(Issue 14) pp:7103-7109
Publication Date(Web):July 9, 2015
DOI:10.1021/acs.inorgchem.5b01204
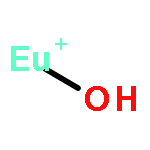
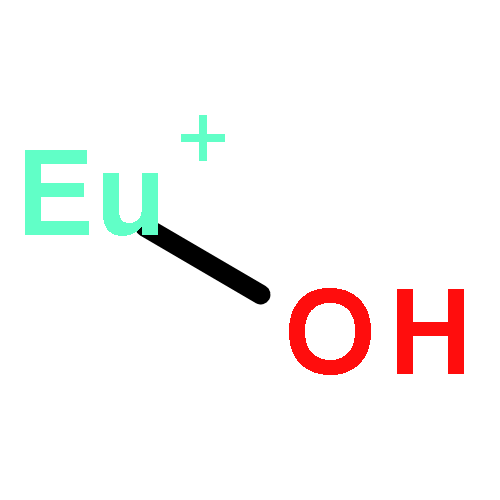
We performed a theoretical investigation for the selectivity of Eu(III)/Am(III) ions depending on the donor atoms by means of all-electron ZORA-DFT calculation. We estimated their selectivity as the relative stability in the complex formation reaction. The B2PLYP functional reproduced the experimental selectivity in which S- and N-donor ligands favor Am(III) ion, but O-donor ligand favors Eu(III) ion. Mulliken’s bond overlap population analysis revealed that the contribution of the f orbital to the bonding was small or zero for Eu complex, whereas it was large for Am complex. The bonding nature of the f orbital for Am ion was the bonding type to S- and N-donor ligands, while it was the antibonding type to O-donor ligand. It was suggested that the difference in the bonding nature between the f orbital in the metal and the donor atoms determines the selectivity of Eu(III)/Am(III) by donor ligands.
Co-reporter:Masashi Kaneko, Sunao Miyashita and Satoru Nakashima
Dalton Transactions 2015 vol. 44(Issue 17) pp:8080-8088
Publication Date(Web):18 Mar 2015
DOI:10.1039/C4DT03064H
We have performed benchmark investigations into the bonding properties in lanthanide and actinide complexes to quantitatively estimate the covalency of f-block compounds. Three different density functionals including BP86 (pure-GGA), B3LYP (hybrid-GGA) and B2PLYP (double hybrid-GGA) were employed for all-electron self-consistent field calculations compensated by the scalar-relativistic zero-order regular approximation (ZORA) Hamiltonian with a relativistically contracted all-electron basis set. Ten Eu and ten Np complexes were employed as benchmark sets for the calculation of Mössbauer parameters for 151Eu and 237Np compounds. As a result of the linear fitting between the calculated electron densities at the nucleus (ρcalc0) and the experimental isomer shifts (δexp), the calculations performed using the all-electron ZORA-B2PLYP level reproduced a change of electron density at the Mössbauer nucleus for both Eu and Np complexes with high correlation coefficients (R2 > 0.90). Mulliken's population analyses indicated that the BP86 and B3LYP methods overestimated the covalency of both Eu and Np complexes due to the smaller amount of the exact Hartree–Fock exchange admixture included in BP86 and B3PLYP compared to that in the B2PLYP functional. By comparing Mulliken's electronic structure analyses with the experimental isomer shifts, we found that Mulliken's spin population values were good parameters to quantitatively estimate the bonding natures of Eu and Np complexes.
Co-reporter:Hiroki Yasuhara, Kazuki Koga, Satoru Nakashima
Journal of Organometallic Chemistry 2015 Volume 779() pp:86-90
Publication Date(Web):1 March 2015
DOI:10.1016/j.jorganchem.2014.12.030
•Biosmocene was synthesized by Ullmann coupling reaction of iodoosmocene.•Biosmocenium-I triiodide salt was obtained by reaction of biosmocene and I2.•The intramolecular electron transfer reaction between OsII and Os IV was observed.•The activation energy of the electron transfer reaction was determined.•The Os–I bond was larger than the Ru–I bond.Selective mono-lithiation method of bis(cyclopentadienyl)osmium(II) (osmocene) was established. Iodoosmocene(C5H4I)Os(C5H5) (OcI), which is the first monohalogenated product of osmocene, was prepared by the reaction of lithioosmocene with I2. Ullmann coupling reaction of iodoosmoene allowed to prepare biosmocene(C5H5)Os(μ2-η5:η5-C10H8)Os(C5H5) (OcOc). OcOc reacted with I2, giving mixed-valence biosmocenium salt [(C5H5)OsII(μ2-η5:η5-C10H8) (C5H5)OsIVI]I3(A). The structure of A was determined by single crystal X-ray structural analysis. The intramolecular electron transfer reaction between OsII and Os IV was observed by using 1H NMR spectroscopy, accompanied by the exchange of I− anion between the two units. The activation energy of the electron transfer reaction was estimated and the value was larger than that of binuclear ruthenocenium salt [(C5H5)RuII(μ2-η5:η5-C10H8) (C5H5)RuIVI]I3(B).Ullmann coupling reaction of iodoosmoene allowed to prepare biosmocene (OcOc). OcOc reacted with I2, giving mixed-valence biosmocenium salt [(C5H5)OsII(μ2-η5:η5-C10H8) (C5H5)OsIVI]I3. The intramolecular electron transfer reaction between OsII and Os IV was observed by using 1H NMR spectroscopy, accompanied by the exchange of I− anion between the two units.
Co-reporter:Masaki Atsuchi, Katsuya Inoue, Satoru Nakashima
Inorganica Chimica Acta 2011 370(1) pp: 82-88
Publication Date(Web):
DOI:10.1016/j.ica.2011.01.028
Co-reporter:Satoru Nakashima, Akiko Yamamoto, Yoritaka Asada, Nobuyoshi Koga, Tsutomu Okuda
Inorganica Chimica Acta 2005 Volume 358(Issue 2) pp:257-264
Publication Date(Web):25 January 2005
DOI:10.1016/j.ica.2004.09.001
Reaction of FeSO4 · 7H2O with trans-1,2-bis(4-pyridyl)ethylene (tvp) and NaNCS in the mixed solvent of water and ethanol gave rise to the formation of different coordination polymers. One (Fe(tvp)2(NCS)2(H2O)2) is hydrogen bonded and π–π interacted structure, while the other ([Fe(tvp)2(NCS)2][0.5(tvp · 2EtOH)]) is 2D grid structure, which enclathrates tvp · 2EtOH. This enclathrated tvp · 2EtOH interacts with the two 2D grid sheets to form 3D structure. The same reaction was carried out with KNCSe instead of NaNCS (Fe(tvp)2(NCSe)2(H2O)2). 57Fe Mössbauer spectra revealed that all the present assembled complexes are in the FeII high-spin state. The dissociation behavior of enclathrated molecule and ligand was investigated by TG, and the resultant electronic state of iron atom was studied by 57Fe Mössbauer spectroscopy.Newly synthesized Fe-tvp-NCS complexes gave two crystal forms, both showed a temperature-independent FeII high-spin state, and the dissociation phenomena were investigated by TG and 57Fe Mössbauer spectroscopy.
Co-reporter:Masashi Kaneko, Sunao Miyashita and Satoru Nakashima
Dalton Transactions 2015 - vol. 44(Issue 17) pp:NaN8088-8088
Publication Date(Web):2015/03/18
DOI:10.1039/C4DT03064H
We have performed benchmark investigations into the bonding properties in lanthanide and actinide complexes to quantitatively estimate the covalency of f-block compounds. Three different density functionals including BP86 (pure-GGA), B3LYP (hybrid-GGA) and B2PLYP (double hybrid-GGA) were employed for all-electron self-consistent field calculations compensated by the scalar-relativistic zero-order regular approximation (ZORA) Hamiltonian with a relativistically contracted all-electron basis set. Ten Eu and ten Np complexes were employed as benchmark sets for the calculation of Mössbauer parameters for 151Eu and 237Np compounds. As a result of the linear fitting between the calculated electron densities at the nucleus (ρcalc0) and the experimental isomer shifts (δexp), the calculations performed using the all-electron ZORA-B2PLYP level reproduced a change of electron density at the Mössbauer nucleus for both Eu and Np complexes with high correlation coefficients (R2 > 0.90). Mulliken's population analyses indicated that the BP86 and B3LYP methods overestimated the covalency of both Eu and Np complexes due to the smaller amount of the exact Hartree–Fock exchange admixture included in BP86 and B3PLYP compared to that in the B2PLYP functional. By comparing Mulliken's electronic structure analyses with the experimental isomer shifts, we found that Mulliken's spin population values were good parameters to quantitatively estimate the bonding natures of Eu and Np complexes.