Co-reporter:Yuya Asahina;Toru Kawakami
Chemical Communications 2017 vol. 53(Issue 13) pp:2114-2117
Publication Date(Web):2017/02/09
DOI:10.1039/C6CC10243C
We developed a one-pot peptide ligation method using two orthogonal thioester precursors and a protecting group for the ligation reaction between Asp and Cys. Combination of the two precursors facilitated the one-pot operation and yielded the entire polypeptide. The usefulness of this method was successfully demonstrated by the total synthesis of histone H4.
Co-reporter:Hironobu Hojo, Toru Kawakami, Yuta Hiroyama, Saburo Aimoto
Tetrahedron Letters 2017 Volume 58, Issue 49(Issue 49) pp:
Publication Date(Web):6 December 2017
DOI:10.1016/j.tetlet.2017.10.074
•An N-alkoxy or N-aryloxyamino group at the N-terminus of a peptide is selectively acylated under acidic condition.•As an acylating agent, peptide thioester with silver ion activation can be used.•N-Methoxy group can be cleaved by samarium(II) iodide treatment.•Using this method, human atrial natriuretic peptide was successfully synthesized.Peptides with an N-alkoxy or N-aryloxy amino acid at their N-terminus were synthesized and successfully ligated with a peptide thioester by silver ion activation under a slightly acidic condition without requiring protection of the side chain amino groups. The N-methoxy group was easily cleaved by the SmI2 reduction in CH3OH aq. to obtain the desired peptide with a native peptide bond. This method was successfully applied to the synthesis of the human atrial natriuretic peptide showing the efficiency of the novel ligation.Download high-res image (91KB)Download full-size image
Co-reporter:Toshiki Takei;Tomoshige Andoh; Dr. Toshifumi Takao; Dr. Hironobu Hojo
Angewandte Chemie International Edition 2017 Volume 56(Issue 49) pp:15708-15711
Publication Date(Web):2017/12/04
DOI:10.1002/anie.201709418
AbstractThe synthesis of a peptide selenoester was efficiently carried out by the 9-fluorenylmethoxycarbonyl (Fmoc) method using N-alkylcysteine, at the C-terminus of the peptide, as the N-to-S acyl shift device. The selenoester selectively reacted with the terminal amino group of the peptide aryl thioester in the presence of N,N-diisopropylethylamine and dipyridyldisulfide, thus leaving the aryl thioester intact. Combined with silver-ion-promoted and silver-ion-free thioester activation methods, a one-pot four-segment ligation was realized. The method was successfully used to assemble the entire sequence of superoxide dismutase (SOD), which is composed of 153 amino-acid residues, in one pot. After the folding reaction, the fully active SOD was obtained.
Co-reporter:Dr. Kenta Arai;Toshiki Takei;Dr. Masaki Okumura;Dr. Satoshi Watanabe;Dr. Yuta Amagai;Dr. Yuya Asahina; Dr. Luis Moroder; Dr. Hironobu Hojo; Dr. Kenji Inaba; Dr. Michio Iwaoka
Angewandte Chemie International Edition 2017 Volume 56(Issue 20) pp:5522-5526
Publication Date(Web):2017/05/08
DOI:10.1002/anie.201701654
AbstractSynthetic insulin analogues with a long lifetime are current drug targets for the therapy of diabetic patients. The replacement of the interchain disulfide with a diselenide bridge, which is more resistant to reduction and internal bond rotation, can enhance the lifetime of insulin in the presence of the insulin-degrading enzyme (IDE) without impairing the hormonal function. The [C7UA,C7UB] variant of bovine pancreatic insulin (BPIns) was successfully prepared by using two selenocysteine peptides (i.e., the C7U analogues of A- and B-chains, respectively). In a buffer solution at pH 10 they spontaneously assembled under thermodynamic control to the correct insulin fold. The selenoinsulin (Se-Ins) exhibited a bioactivity comparable to that of BPIns. Interestingly, degradation of Se-Ins with IDE was significantly decelerated (τ1/2≈8 h vs. ≈1 h for BPIns). The lifetime enhancement could be due to both the intrinsic stability of the diselenide bond and local conformational changes induced by the substitution.
Co-reporter:Dr. Kenta Arai;Toshiki Takei;Dr. Masaki Okumura;Dr. Satoshi Watanabe;Dr. Yuta Amagai;Dr. Yuya Asahina; Dr. Luis Moroder; Dr. Hironobu Hojo; Dr. Kenji Inaba; Dr. Michio Iwaoka
Angewandte Chemie 2017 Volume 129(Issue 20) pp:5614-5618
Publication Date(Web):2017/05/08
DOI:10.1002/ange.201701654
AbstractSynthetic insulin analogues with a long lifetime are current drug targets for the therapy of diabetic patients. The replacement of the interchain disulfide with a diselenide bridge, which is more resistant to reduction and internal bond rotation, can enhance the lifetime of insulin in the presence of the insulin-degrading enzyme (IDE) without impairing the hormonal function. The [C7UA,C7UB] variant of bovine pancreatic insulin (BPIns) was successfully prepared by using two selenocysteine peptides (i.e., the C7U analogues of A- and B-chains, respectively). In a buffer solution at pH 10 they spontaneously assembled under thermodynamic control to the correct insulin fold. The selenoinsulin (Se-Ins) exhibited a bioactivity comparable to that of BPIns. Interestingly, degradation of Se-Ins with IDE was significantly decelerated (τ1/2≈8 h vs. ≈1 h for BPIns). The lifetime enhancement could be due to both the intrinsic stability of the diselenide bond and local conformational changes induced by the substitution.
Co-reporter:Toshiki Takei;Tomoshige Andoh; Dr. Toshifumi Takao; Dr. Hironobu Hojo
Angewandte Chemie 2017 Volume 129(Issue 49) pp:15914-15917
Publication Date(Web):2017/12/04
DOI:10.1002/ange.201709418
AbstractThe synthesis of a peptide selenoester was efficiently carried out by the 9-fluorenylmethoxycarbonyl (Fmoc) method using N-alkylcysteine, at the C-terminus of the peptide, as the N-to-S acyl shift device. The selenoester selectively reacted with the terminal amino group of the peptide aryl thioester in the presence of N,N-diisopropylethylamine and dipyridyldisulfide, thus leaving the aryl thioester intact. Combined with silver-ion-promoted and silver-ion-free thioester activation methods, a one-pot four-segment ligation was realized. The method was successfully used to assemble the entire sequence of superoxide dismutase (SOD), which is composed of 153 amino-acid residues, in one pot. After the folding reaction, the fully active SOD was obtained.
Co-reporter:Yuya Asahina, Kei Nabeshima, Hironobu Hojo
Tetrahedron Letters 2015 Volume 56(Issue 11) pp:1370-1373
Publication Date(Web):11 March 2015
DOI:10.1016/j.tetlet.2015.01.095
Peptides having the C-terminal N-alkylcysteine (NAC) with a free carboxy group, which can be easily prepared by the conventional 9-fluorenylmethoxycarbonyl (Fmoc) solid-phase peptide synthesis (SPPS), was directly used for the native chemical ligation (NCL) based on the in situ thioesterification method. The reaction efficiently proceeded under a mild acidic condition (pH ∼5) to give the ligated product. This method was successfully used for the synthesis of the human brain natriuretic peptide, (BNP)-32, showing the usefulness of the peptidyl NAC as a thioester surrogate for the NCL reaction.
Co-reporter:Dr. Yuya Asahina;Shinobu Komiya;Ami Ohagi;Rina Fujimoto;Dr. Hiroko Tamagaki;Katsuhiro Nakagawa;Dr. Takashi Sato;Dr. Shizuo Akira;Dr. Toshifumi Takao;Dr. Akira Ishii;Dr. Yoshiaki Nakahara;Dr. Hironobu Hojo
Angewandte Chemie International Edition 2015 Volume 54( Issue 28) pp:8226-8230
Publication Date(Web):
DOI:10.1002/anie.201501847
Abstract
The chemical synthesis of human interleukin-2 (IL-2) , having a core 1 sugar, by a ligation method is reported. Although IL-2 is a globular glycoprotein, its C-terminal region, in particular (99-133), is extremely insoluble when synthesized by solid-phase method. To overcome this problem, the side-chain carboxylic acid of the Glu residues was protected by a picolyl ester, thus reversing its polarity from negative to positive. This reverse polarity protection significantly increased the isoelectric point of the peptide segment and made it positive under acidic conditions and facilitated the purification. An efficient method to prepare the prolyl peptide thioester required for the synthesis of the (28-65) segment was also developed. These efforts resulted in the total synthesis of the glycosylated IL-2 having full biological activity.
Co-reporter:Dr. Yuya Asahina;Shinobu Komiya;Ami Ohagi;Rina Fujimoto;Dr. Hiroko Tamagaki;Katsuhiro Nakagawa;Dr. Takashi Sato;Dr. Shizuo Akira;Dr. Toshifumi Takao;Dr. Akira Ishii;Dr. Yoshiaki Nakahara;Dr. Hironobu Hojo
Angewandte Chemie 2015 Volume 127( Issue 28) pp:8344-8348
Publication Date(Web):
DOI:10.1002/ange.201501847
Abstract
The chemical synthesis of human interleukin-2 (IL-2) , having a core 1 sugar, by a ligation method is reported. Although IL-2 is a globular glycoprotein, its C-terminal region, in particular (99-133), is extremely insoluble when synthesized by solid-phase method. To overcome this problem, the side-chain carboxylic acid of the Glu residues was protected by a picolyl ester, thus reversing its polarity from negative to positive. This reverse polarity protection significantly increased the isoelectric point of the peptide segment and made it positive under acidic conditions and facilitated the purification. An efficient method to prepare the prolyl peptide thioester required for the synthesis of the (28-65) segment was also developed. These efforts resulted in the total synthesis of the glycosylated IL-2 having full biological activity.
Co-reporter:Toshiki Takei, Yoshiko Urabe, Yuya Asahina, Hironobu Hojo, Takeshi Nomura, Kenichi Dedachi, Kenta Arai, and Michio Iwaoka
The Journal of Physical Chemistry B 2014 Volume 118(Issue 2) pp:492-500
Publication Date(Web):December 19, 2013
DOI:10.1021/jp4113975
Although the catalytic triad of glutathione peroxidase (GPx) has been well recognized, there was little evidence for the relevance of the interactions among the triad amino acid residues, i.e., selenocysteine (U), glutamine (Q), and tryptophan (W), to the GPx antioxidative functions. Using a designed selenopeptide having an amino acid sequence of GQAUAWG, we demonstrate here that U, Q, and W present at the active site can interact with each other to exert the enzymatic activity. The amino acid sequence was chosen on the basis of the Monte Carlo molecular simulation for various selenopeptides in polarizable continuous water using the SAAP force field (SAAP-MC). Measurement of the GPx-like activity for the selenopeptide obtained by solid-phase peptide synthesis revealed that the antioxidant activity is cooperatively enhanced by the presence of Q and W proximate to U, although the activity was low compared to selenocystine (U2). The effect of Q on the activity was more important than that of W. In addition, the fluorescence spectrometry suggested a close contact between U and W. These experimental observations were supported by SAAP-MC simulation as well as by ab initio calculation. The latter further suggested that the interaction mode among the triad changes depending on the intermediate states.
Co-reporter:Yuya Asahina, Toru Kawakami and Hironobu Hojo
Chemical Communications 2017 - vol. 53(Issue 13) pp:NaN2117-2117
Publication Date(Web):2017/01/24
DOI:10.1039/C6CC10243C
We developed a one-pot peptide ligation method using two orthogonal thioester precursors and a protecting group for the ligation reaction between Asp and Cys. Combination of the two precursors facilitated the one-pot operation and yielded the entire polypeptide. The usefulness of this method was successfully demonstrated by the total synthesis of histone H4.

![L-Glutamic acid, N-[(9H-fluoren-9-ylmethoxy)carbonyl]-, 1-(1H-benzotriazol-1-yl) 5-(4-pyridinylmethyl) ester](/data/chemimg/3742000/1800569-83-8.png)
![L-Glutamic acid, N-[(9H-fluoren-9-ylmethoxy)carbonyl]-, 1-(1H-benzotriazol-1-yl) 5-(4-pyridinylmethyl) ester](/data/chemimg/3742000/1800569-83-8_b.png)
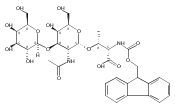
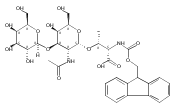
![L-Threonine, O-[N-[(9H-fluoren-9-ylmethoxy)carbonyl]-L-alanyl]-N-[(4-pyridinylmethoxy)carbonyl]-](/data/chemimg/3742000/1792229-65-2.png)
![L-Threonine, O-[N-[(9H-fluoren-9-ylmethoxy)carbonyl]-L-alanyl]-N-[(4-pyridinylmethoxy)carbonyl]-](/data/chemimg/3742000/1792229-65-2_b.png)
![L-Threonine, O-[N-[(9H-fluoren-9-ylmethoxy)carbonyl]-L-alanyl]-N-[(4-pyridinylmethoxy)carbonyl]-, 1,1-dimethylethyl ester](/data/chemimg/3742000/1792229-64-1.png)
![L-Threonine, O-[N-[(9H-fluoren-9-ylmethoxy)carbonyl]-L-alanyl]-N-[(4-pyridinylmethoxy)carbonyl]-, 1,1-dimethylethyl ester](/data/chemimg/3742000/1792229-64-1_b.png)
![L-Glutamic acid, N-[(9H-fluoren-9-ylmethoxy)carbonyl]-, 1-(1,1-dimethylethyl) 5-(4-pyridinylmethyl) ester](/data/chemimg/3742000/1564250-96-9.png)
![L-Glutamic acid, N-[(9H-fluoren-9-ylmethoxy)carbonyl]-, 1-(1,1-dimethylethyl) 5-(4-pyridinylmethyl) ester](/data/chemimg/3742000/1564250-96-9_b.png)
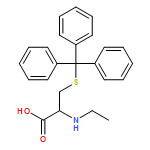
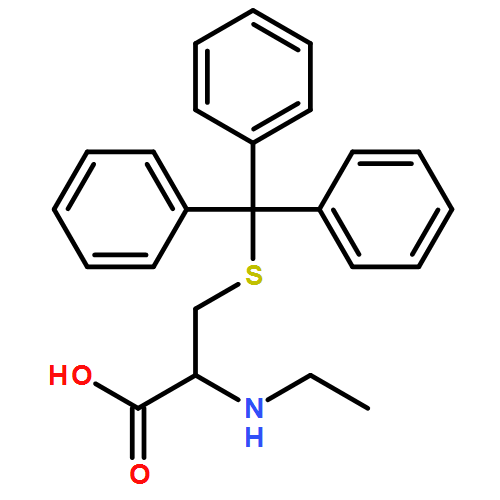
![L-Cysteine, 1-[(9H-fluoren-9-ylmethoxy)carbonyl]-L-prolyl-N-ethyl-S-(triphenylmethyl)-](/data/chemimg/3742000/1334511-54-4.png)
![L-Cysteine, 1-[(9H-fluoren-9-ylmethoxy)carbonyl]-L-prolyl-N-ethyl-S-(triphenylmethyl)-](/data/chemimg/3742000/1334511-54-4_b.png)
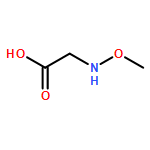
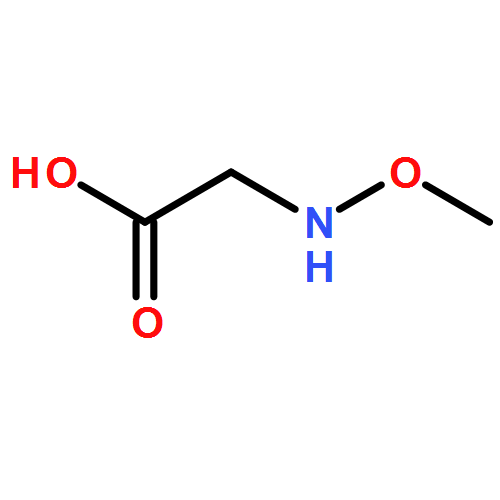
![Glycine, N-[(1,1-dimethylethoxy)carbonyl]-N-methoxy-, 2,3,4,5,6-pentafluorophenyl ester](/data/chemimg/3741400/916460-24-7.png)
![Glycine, N-[(1,1-dimethylethoxy)carbonyl]-N-methoxy-, 2,3,4,5,6-pentafluorophenyl ester](/data/chemimg/3741400/916460-24-7_b.png)
![L-Threonine, N-[(4-pyridinylmethoxy)carbonyl]-, 1,1-dimethylethyl ester](/data/chemimg/3742000/1459694-91-7.png)
![L-Threonine, N-[(4-pyridinylmethoxy)carbonyl]-, 1,1-dimethylethyl ester](/data/chemimg/3742000/1459694-91-7_b.png)




![b-D-Galactopyranose,2-azido-2-deoxy-1-O-[(1,1-dimethylethyl)diphenylsilyl]-4,6-O-[(S)-phenylmethylene]-(9CI)](http://img.cochemist.com/ccimg/132200/132183-16-5.png)
![b-D-Galactopyranose,2-azido-2-deoxy-1-O-[(1,1-dimethylethyl)diphenylsilyl]-4,6-O-[(S)-phenylmethylene]-(9CI)](http://img.cochemist.com/ccimg/132200/132183-16-5_b.png)
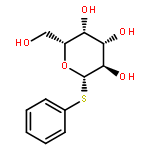
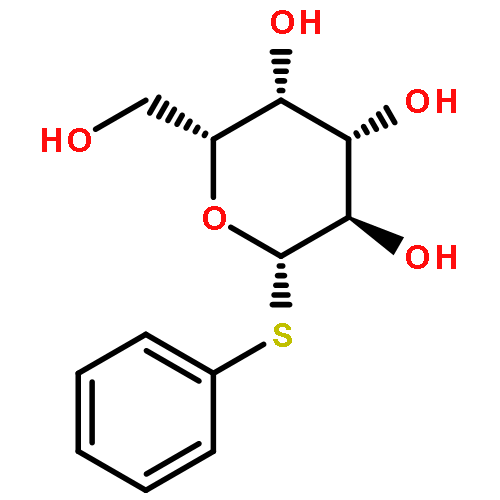
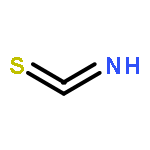
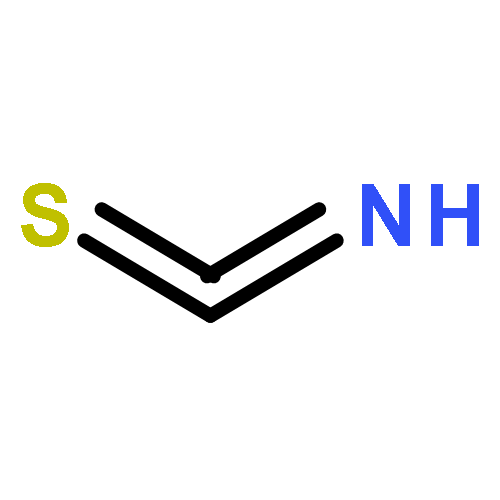
![L-Lysine, N2-[(9H-fluoren-9-ylmethoxy)carbonyl]-N6-[(4-pyridinylmethoxy)carbonyl]-, 1H-benzotriazol-1-yl ester](/data/chemimg/3742000/1644305-24-7.png)
![L-Lysine, N2-[(9H-fluoren-9-ylmethoxy)carbonyl]-N6-[(4-pyridinylmethoxy)carbonyl]-, 1H-benzotriazol-1-yl ester](/data/chemimg/3742000/1644305-24-7_b.png)