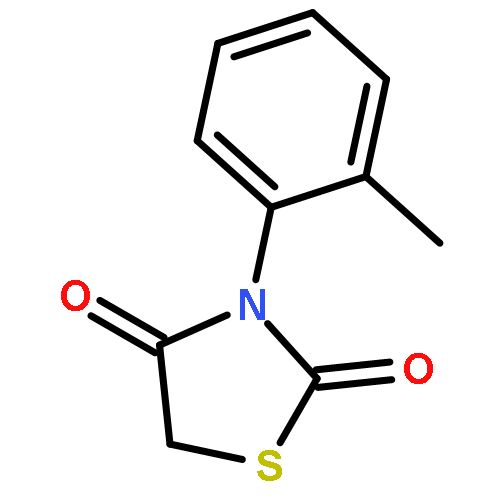
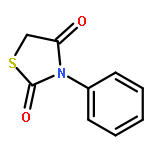
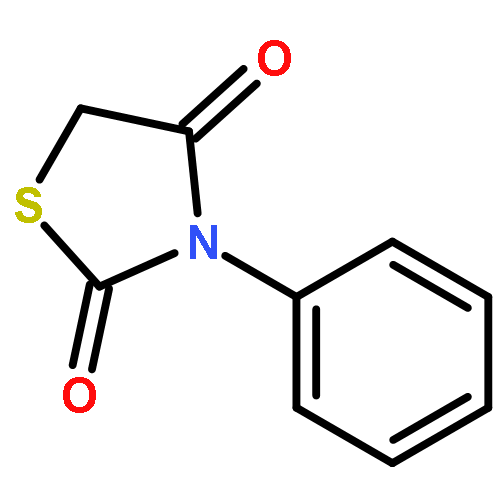
Neuronal cell death is the main cause behind the progressive loss of brain function in age-related neurodegenerative disorders such as Alzheimer’s and Parkinson’s diseases. Despite the differing etiologies of these neurological diseases, the underlying neuronal damage is triggered by common mechanisms such as oxidative stress, impaired calcium homeostasis, and disrupted mitochondrial integrity and function. In particular, mitochondrial fragmentation, mitochondrial membrane permeability, and the release of death-promoting factors into the cytosol have been revealed as the “point of no return” in programmed cell death in neurons. Recent studies revealed a pivotal role for the pro-apoptotic Bcl-2-family protein Bid in models of neuronal cell death, which confirmed Bid as a potential drug target. Herein, we present N-acyl-substituted derivatives of 4-phenoxyaniline that were screened for their potential to attenuate Bid-mediated neurotoxicity. These compounds provided significant protection against glutamate- and Bid-induced toxicity in cultured neurons. Substitution of the amino group in the 4-phenoxyaniline scaffold with 4-piperidine carboxylic acid and N-hydroxyethyl-4-piperidine carboxylic acid yielded compounds that displayed significant neuroprotective activity at concentrations as low as 1 μM. Furthermore, findings of a tBid-overexpression assay and real-time measurements of cell impedance support the hypothesis that these compounds indeed address the Bid protein.
Malaria is a potentially fatal disease caused by Plasmodium parasites and poses a major medical risk in large parts of the world. The development of new, affordable antimalarial drugs is of vital importance as there are increasing reports of resistance to the currently available therapeutics. In addition, most of the current drugs used for chemoprophylaxis merely act on parasites already replicating in the blood. At this point, a patient might already be suffering from the symptoms associated with the disease and could additionally be infectious to an Anopheles mosquito. These insects act as a vector, subsequently spreading the disease to other humans. In order to cure not only malaria but prevent transmission as well, a drug must target both the blood- and pre-erythrocytic liver stages of the parasite. P. falciparum (Pf) enoyl acyl carrier protein (ACP) reductase (ENR) is a key enzyme of plasmodial type II fatty acid biosynthesis (FAS II). It has been shown to be essential for liver-stage development of Plasmodium berghei and is therefore qualified as a target for true causal chemoprophylaxis. Using virtual screening based on two crystal structures of PfENR, we identified a structurally novel class of FAS inhibitors. Subsequent chemical optimization yielded two compounds that are effective against multiple stages of the malaria parasite. These two most promising derivatives were found to inhibit blood-stage parasite growth with IC50 values of 1.7 and 3.0 μM and lead to a more prominent developmental attenuation of liver-stage parasites than the gold-standard drug, primaquine.
1-Deoxy-D-xylulose-5-phosphate reductoisomerase (Dxr) represents an essential enzyme of the mevalonate-independent pathway of the isoprenoid biosynthesis. Using fosmidomycin as a specific inhibitor of Dxr, this enzyme was previously validated as target for the treatment of malaria and bacterial infections. The replacement of the formyl residue of fosmidomycin by spacious acyl residues yielded inhibitors active in the micromolar range. As predicted by flexible docking, evidence was obtained for the formation of a hydrogen bond between an appropriately placed carbonyl group in the acyl residue and the main-chain NH of Met214 located in the flexible catalytic loop of the enzyme.
Fosmidomycin and FR900098 are inhibitors of the 1-deoxy-D-xylulose 5-phosphate reductoisomerase (DXR; IspC), a key enzyme of the mevalonate-independent isoprenoid biosynthesis pathway. We have determined the in-vitro antimalarial activity of two double ester prodrugs 2, 3 in direct comparison with the unmodified FR900098 1 against intraerythrocytic forms of Plasmodium falciparum. Temporarily masking the polar properties of the phosphonate moiety of the DXR inhibitor FR900098 1 enhanced not only its oral bioavailability but also the intrinsic activity of this series against the parasites.
Since ancient times, humankind has had to struggle against the persistent onslaught of pathogenic microorganisms. Nowadays, malaria is still the most important infectious disease worldwide. Considerable success in gaining control over malaria was achieved in the 1950s and 60s through landscaping measures, vector control with the insecticide DDT, and the widespread administration of chloroquine, the most important antimalarial agent ever. In the late 1960s, the final victory over malaria was believed to be within reach. However, the parasites could not be eradicated because they developed resistance against the most widely used and affordable drugs of that time. Today, cases of malaria infections are on the rise and have reached record numbers. This review gives a short description of the malaria disease, briefly addresses the history of antimalarial drug development, and focuses on drugs currently available for malaria therapy. The present knowledge regarding their mode of action and the mechanisms of resistance are explained, as are the attempts made by numerous research groups to overcome the resistance problem within classes of existing drugs and in some novel classes. Finally, this review covers all classes of antimalarials for which at least one drug candidate is in clinical development. Antimalarial agents that are solely in early development stages will be addressed in a separate review.







![Thiophene, 2-[1,1'-biphenyl]-4-yl-](http://img.cochemist.com/ccimg/38000/37909-88-9.png)
![Thiophene, 2-[1,1'-biphenyl]-4-yl-](http://img.cochemist.com/ccimg/38000/37909-88-9_b.png)