Co-reporter:Eric C. Truong, Puay W. Phuan, Amanda L. Reggi, Loretta Ferrera, Luis J. V. Galietta, Sarah E. Levy, Alannah C. Moises, Onur Cil, Elena Diez-Cecilia, Sujin Lee, Alan S. Verkman, and Marc O. Anderson
Journal of Medicinal Chemistry June 8, 2017 Volume 60(Issue 11) pp:4626-4626
Publication Date(Web):May 11, 2017
DOI:10.1021/acs.jmedchem.7b00020
Transmembrane protein 16A (TMEM16A), also called anoctamin 1 (ANO1), is a calcium-activated chloride channel expressed widely mammalian cells, including epithelia, vascular smooth muscle tissue, electrically excitable cells, and some tumors. TMEM16A inhibitors have been proposed for treatment of disorders of epithelial fluid and mucus secretion, hypertension, asthma, and possibly cancer. Herein we report, by screening, the discovery of 2-acylaminocycloalkylthiophene-3-carboxylic acid arylamides (AACTs) as inhibitors of TMEM16A and analysis of 48 synthesized analogs (10ab–10bw) of the original AACT compound (10aa). Structure–activity studies indicated the importance of benzene substituted as 2- or 4-methyl, or 4-fluoro, and defined the significance of thiophene substituents and size of the cycloalkylthiophene core. The most potent compound (10bm), which contains an unusual bromodifluoroacetamide at the thiophene 2-position, had IC50 of ∼30 nM, ∼3.6-fold more potent than the most potent previously reported TMEM16A inhibitor 4 (Ani9), and >10-fold improved metabolic stability. Direct and reversible inhibition of TMEM16A by 10bm was demonstrated by patch-clamp analysis. AACTs may be useful as pharmacological tools to study TMEM16A function and as potential drug development candidates.
Co-reporter:Scott Eagon ;Marc O. Anderson
European Journal of Organic Chemistry 2014 Volume 2014( Issue 8) pp:1653-1665
Publication Date(Web):
DOI:10.1002/ejoc.201301580
Abstract
A microwave-mediated Pictet–Spengler procedure utilizing 1,2-dichloroethane (DCE) and trifluoroacetic acid (TFA) was developed to provide tetrahydro-β-carboline salts in high yields. Reactions are complete in 20 minutes or less and the product precipitates from solution in high yields and purity, negating the need for liquid–liquid extraction or column chromatography. This method tolerates a wide range of functionality and can be performed on milligram to gram scales. A subsequent microwave-mediated aromatization of the synthesized tetrahydro-β-carbolines to β-carbolines was also developed utilizing catalytic Pd/C. The aromatization is complete in 60 min or less with most substrates requiring minimal purification.
Co-reporter:Y. Liu, C. Esteva-Font, C. Yao, P.W. Phuan, A.S. Verkman, M.O. Anderson
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 11) pp:3338-3341
Publication Date(Web):1 June 2013
DOI:10.1016/j.bmcl.2013.03.089
The kidney urea transport protein UT-B is an attractive target for the development of small-molecule inhibitors with a novel diuretic (‘urearetic’) action. Previously, two compounds in the triazolothienopyrimidine scaffold (1a and 1c) were reported as UT-B inhibitors. Compound 1c incorporates a 1,1-difluoroethyl group, which affords improved microsomal stability when compared to the corresponding ethyl-substituted compound 1a. Here, a small focused library (4a–4f) was developed around lead inhibitor 1c to investigate the requirement of an amidine-linked thiophene in the inhibitor scaffold. Two compounds (4a and 4b) with nanomolar inhibitory potency (IC50 ≈ 40 nM) were synthesized. Computational docking of lead structure 1c and 4a–4f into a homology model of the UT-B cytoplasmic surface suggested binding with the core heterocycle buried deep into the hydrophobic pore region of the protein.
Co-reporter:Marc O. Anderson ; Jicheng Zhang ; Yan Liu ; Chenjuan Yao ; Puay-Wah Phuan ;A. S. Verkman
Journal of Medicinal Chemistry 2012 Volume 55(Issue 12) pp:5942-5950
Publication Date(Web):June 13, 2012
DOI:10.1021/jm300491y
Urea transporters, which include UT-B in kidney microvessels, are potential targets for development of drugs with a novel diuretic (‘urearetic’) mechanism. We recently identified, by high-throughput screening, a triazolothienopyrimidine UT-B inhibitor, 1, that selectively and reversibly inhibited urea transport with IC50 = 25.1 nM and reduced urinary concentration in mice (Yao et al. J. Am. Soc. Nephrol., in press). Here, we analyzed 273 commercially available analogues of 1 to establish a structure–activity series and synthesized a targeted library of 11 analogues to identify potent, metabolically stable UT-B inhibitors. The best compound, {3-[4-(1,1-difluoroethyl)benzenesulfonyl]thieno[2,3-e][1,2,3]triazolo[1,5-a]pyrimidin-5-yl}thiophen-2-ylmethylamine, 3k, had IC50 of 23 and 15 nM for inhibition of urea transport by mouse and human UT-B, respectively, and ∼40-fold improved in vitro metabolic stability compared to 1. In mice, 3k accumulated in kidney and urine and reduced maximum urinary concentration. Triazolothienopyrimidines may be useful for therapy of diuretic-refractory edema in heart and liver failure.
Co-reporter:Brian R. Blank;Pinar Alayoglu;William Engen;Joseph K. Choi;Clifford E. Berkman;Marc O. Anderson
Chemical Biology & Drug Design 2011 Volume 77( Issue 4) pp:241-247
Publication Date(Web):
DOI:10.1111/j.1747-0285.2011.01085.x
Glutamate carboxypeptidase II (GCP2) is a membrane-bound cell-surface peptidase which is implicated in several neurological disorders and is also over-expressed in prostate tumor cells. There is a significant interest in the inhibition of GCP2 as a means of neuroprotection, while GCP2 inhibition as a method to treat prostate cancer remains a topic of further investigation. The key zinc-binding functional group of the well-characterized classes of GCP2 inhibitors (phosphonates and phosphoramidates) is tetrahedral and negatively charged at neutral pH, while glutamyl urea class of inhibitors possesses a planar and neutral zinc-binding group. This study introduces a new class of GCP2 inhibitors, N-substituted glutamyl sulfonamides, which possess a neutral tetrahedral zinc-binding motif. A library containing 15 secondary sulfonamides and 4 tertiary (N-methyl) sulfonamides was prepared and evaluated for inhibitory potency against purified GCP2 enzyme activity. While most inhibitors lacked potency at 100 μm, short alkyl sulfonamides exhibited promising low micromolar potency, with the optimal inhibitor in this series being glutamyl N-(propylsulfonamide) (2g). Lastly, molecular docking was used to develop a model to formulate an explanation for the relative inhibitory potencies employed for this class of inhibitors.
Co-reporter:William Engen, Terrence E. O’Brien, Brendan Kelly, Jacinda Do, Liezel Rillera, Lance K. Stapleton, Jack F. Youngren, Marc O. Anderson
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 16) pp:5995-6005
Publication Date(Web):15 August 2010
DOI:10.1016/j.bmc.2010.06.071
The insulin-like growth factor receptor (IGF-1R) is a receptor tyrosine kinase (RTK) involved in all stages of the development and propagation of breast and other cancers. The inhibition of IGF-1R by small molecules remains a promising strategy to treat cancer. Herein, we explore SAR around previously characterized lead compound (1), which is an aryl-heteroaryl urea (AHU) consisting of 4-aminoquinaldine and a substituted aromatic ring system. A library of novel AHU compounds was prepared based on derivatives of the 4-aminoquinoline heterocycle (including various 2-substituted derivatives, and naphthyridines). The compounds were screened for in vitro inhibitory activity against IGF-1R, and several compounds with improved activity (3–5 μM) were identified. Furthermore, a computational docking study was performed, which identifies a fairly consistent lowest energy mode of binding for the more-active set of inhibitors in this series, while the less-active inhibitors do not adopt a consistent mode of binding.

![N-[4-(1-benzofuran-2-yl)-1,3-thiazol-2-yl]-2-chloroacetamide](http://img.cochemist.com/ccimg/924200/924129-01-1.png)
![N-[4-(1-benzofuran-2-yl)-1,3-thiazol-2-yl]-2-chloroacetamide](http://img.cochemist.com/ccimg/924200/924129-01-1_b.png)
![2-bromo-n-[4-(4-chlorophenyl)-1,3-thiazol-2-yl]acetamide](/data/chemimg/54600/448224-88-2.png)
![2-bromo-n-[4-(4-chlorophenyl)-1,3-thiazol-2-yl]acetamide](/data/chemimg/54600/448224-88-2_b.png)
![2-chloro-N-[4-(2-thienyl)-1,3-thiazol-2-yl]acetamide](http://img.cochemist.com/ccimg/436100/436094-95-0.png)
![2-chloro-N-[4-(2-thienyl)-1,3-thiazol-2-yl]acetamide](http://img.cochemist.com/ccimg/436100/436094-95-0_b.png)
![1-[4-(TRIFLUOROMETHYL)PHENYL]-2,3,4,9-TETRAHYDRO-1H-BETA-CARBOLINE HYDROCHLORIDE](http://img.cochemist.com/ccimg/367300/367251-31-8.png)
![1-[4-(TRIFLUOROMETHYL)PHENYL]-2,3,4,9-TETRAHYDRO-1H-BETA-CARBOLINE HYDROCHLORIDE](http://img.cochemist.com/ccimg/367300/367251-31-8_b.png)
![9H-Pyrido[3,4-b]indole, 1-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/139700/139655-06-4.png)
![9H-Pyrido[3,4-b]indole, 1-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/139700/139655-06-4_b.png)





![9H-Pyrido[3,4-b]indole-3-carboxylicacid, 1-methyl-](http://img.cochemist.com/ccimg/22400/22329-38-0.png)
![9H-Pyrido[3,4-b]indole-3-carboxylicacid, 1-methyl-](http://img.cochemist.com/ccimg/22400/22329-38-0_b.png)
![9H-Pyrido[3,4-b]indole, 1-(1-methylethyl)-](http://img.cochemist.com/ccimg/22400/22314-95-0.png)
![9H-Pyrido[3,4-b]indole, 1-(1-methylethyl)-](http://img.cochemist.com/ccimg/22400/22314-95-0_b.png)
![1-ethyl-9H-Pyrido[3,4-b]indole](http://img.cochemist.com/ccimg/20200/20127-61-1.png)
![1-ethyl-9H-Pyrido[3,4-b]indole](http://img.cochemist.com/ccimg/20200/20127-61-1_b.png)
![Acetamide,2-chloro-N-[4-(1H-indol-3-yl)-2-thiazolyl]-](http://img.cochemist.com/ccimg/19800/19750-29-9.png)
![Acetamide,2-chloro-N-[4-(1H-indol-3-yl)-2-thiazolyl]-](http://img.cochemist.com/ccimg/19800/19750-29-9_b.png)
![1-phenyl-9H-Pyrido[3,4-b]indole](http://img.cochemist.com/ccimg/16800/16765-79-0.png)
![1-phenyl-9H-Pyrido[3,4-b]indole](http://img.cochemist.com/ccimg/16800/16765-79-0_b.png)
![9H-Pyrido[3,4-b]indole-3-carboxylic acid, 1-methyl-, methyl ester](/data/chemimg/924600/16641-82-0.png)
![9H-Pyrido[3,4-b]indole-3-carboxylic acid, 1-methyl-, methyl ester](/data/chemimg/924600/16641-82-0_b.png)
![1H-Pyrido[3,4-b]indole, 2,3,4,9-tetrahydro-1-methyl-,monohydrochloride](http://img.cochemist.com/ccimg/6700/6678-82-6.png)
![1H-Pyrido[3,4-b]indole, 2,3,4,9-tetrahydro-1-methyl-,monohydrochloride](http://img.cochemist.com/ccimg/6700/6678-82-6_b.png)
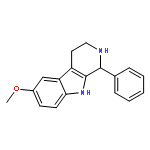
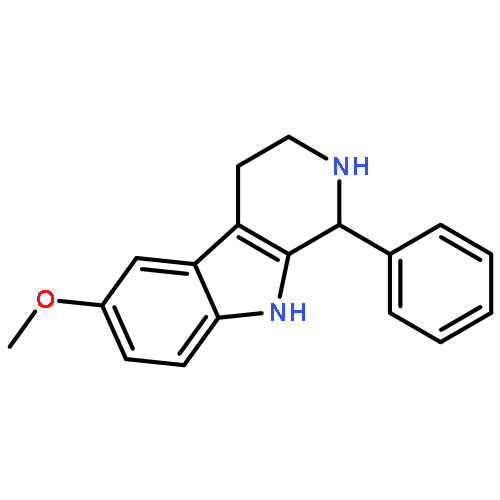






![6-methoxy-1-methyl-9H-Pyrido[3,4-b]indole](http://img.cochemist.com/ccimg/3600/3589-72-8.png)
![6-methoxy-1-methyl-9H-Pyrido[3,4-b]indole](http://img.cochemist.com/ccimg/3600/3589-72-8_b.png)
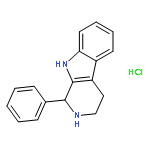
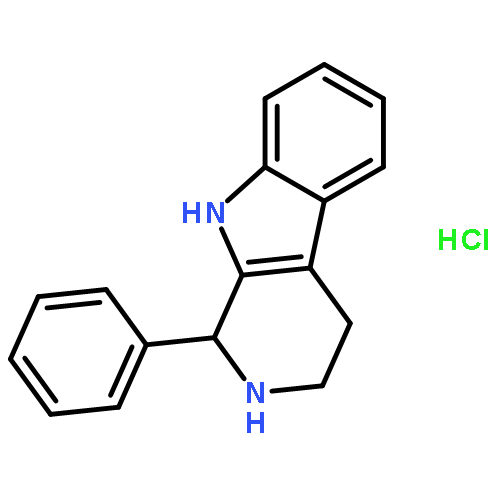


![1-(4-METHOXYPHENYL)-2,3,4,9-TETRAHYDRO-1H-PYRIDO[3,4-B]INDOLE](http://img.cochemist.com/ccimg/3400/3380-73-2.png)
![1-(4-METHOXYPHENYL)-2,3,4,9-TETRAHYDRO-1H-PYRIDO[3,4-B]INDOLE](http://img.cochemist.com/ccimg/3400/3380-73-2_b.png)
![N-{[(2-METHYL-2-PROPANYL)OXY]CARBONYL}-L-LEUCYL-N-[(2S)-5-CARBAMI<WBR />MIDAMIDO-1-OXO-2-PENTANYL]-L-LEUCINAMIDE](http://img.cochemist.com/ccimg/3100/3084-04-6.png)
![N-{[(2-METHYL-2-PROPANYL)OXY]CARBONYL}-L-LEUCYL-N-[(2S)-5-CARBAMI<WBR />MIDAMIDO-1-OXO-2-PENTANYL]-L-LEUCINAMIDE](http://img.cochemist.com/ccimg/3100/3084-04-6_b.png)



![9H-Pyrido[3,4-b]indole,1-methyl-](http://img.cochemist.com/ccimg/500/486-84-0.png)
![9H-Pyrido[3,4-b]indole,1-methyl-](http://img.cochemist.com/ccimg/500/486-84-0_b.png)