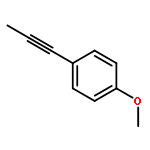
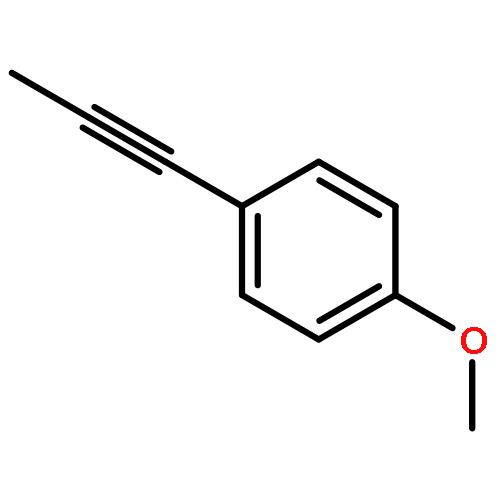
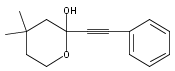
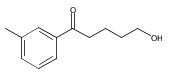
Co-reporter:Xiao-Hui Yang, Alexander Lu, and Vy M. Dong
Journal of the American Chemical Society October 11, 2017 Volume 139(Issue 40) pp:14049-14049
Publication Date(Web):September 27, 2017
DOI:10.1021/jacs.7b09188
We report a Rh-catalyzed hydroamination of 1,3-dienes to generate homoallylic amines. Our work showcases the first case of anti-Markovnikov selectivity in the intermolecular coupling of amines and 1,3-dienes. By tuning the ligand properties and Brønsted acid additive, we find that a combination of rac-BINAP and mandelic acid is critical for achieving anti-Markovnikov selectivity.
Co-reporter:Faben A. Cruz, Yamin Zhu, Quentin D. Tercenio, Zengming Shen, and Vy M. Dong
Journal of the American Chemical Society August 9, 2017 Volume 139(Issue 31) pp:10641-10641
Publication Date(Web):July 25, 2017
DOI:10.1021/jacs.7b05893
We report an enantioselective coupling between alkynes and indoles. A Rh-hydride catalyst isomerizes alkynes to generate a metal-allyl species that can be trapped with both aromatic and heteroaromatic nucleophiles.
Co-reporter:Daniel K. Kim, Jan Riedel, Raphael S. Kim, and Vy M. Dong
Journal of the American Chemical Society August 2, 2017 Volume 139(Issue 30) pp:10208-10208
Publication Date(Web):July 13, 2017
DOI:10.1021/jacs.7b05327
Over the past 40 years, intramolecular hydroacylation has favored five-membered rings, in preference to four membered rings. Herein, we report a catalyst derived from earth-abundant cobalt that enables preparation of cyclobutanones, with excellent regio-, diastereo-, and enantiocontrol, under mild conditions (2 mol % catalyst loading and as low as 50 °C).
Co-reporter:Faben A. Cruz and Vy M. Dong
Journal of the American Chemical Society 2017 Volume 139(Issue 3) pp:1029-1032
Publication Date(Web):January 11, 2017
DOI:10.1021/jacs.6b10680
We report an enantioselective coupling between α-branched aldehydes and alkynes to generate vicinal quaternary and tertiary carbon stereocenters. The choice of Rh and organocatalyst combination allows for access to all possible stereoisomers with high enantio-, diastereo-, and regioselectivity. Our study highlights the power of catalysis to activate two common functional groups and provide access to divergent stereoisomers and constitutional structures.
Co-reporter:Xiao-Hui Yang and Vy M. Dong
Journal of the American Chemical Society 2017 Volume 139(Issue 5) pp:1774-1777
Publication Date(Web):January 27, 2017
DOI:10.1021/jacs.6b12307
We communicate a strategy for the hydrofunctionalization of 1,3-dienes via Rh-hydride catalysis. Conjugated dienes are coupled to nucleophiles to demonstrate the feasibility of novel C–C, C–O, C–S, and C–N bond forming processes. In the presence of a chiral JoSPOphos ligand, hydroamination generates chiral allylic amines with high regio- and enantioselectivity. Tuning both the pKa and steric properties of an acid-additive is critical for enantiocontrol.
Co-reporter:Diane N. Le, Jan Riedel, Natalia Kozlyuk, Rachel W. MartinVy M. Dong
Organic Letters 2017 Volume 19(Issue 1) pp:114-117
Publication Date(Web):December 14, 2016
DOI:10.1021/acs.orglett.6b03308
Dehydrophenylalanine is used as a traceless turn-inducer in the total synthesis of dichotomin E. Macrocyclization of the monomer is achieved in high yields and selectivity over cyclodimerization under conditions 100 times more concentrated than previously achieved. The enamide facilitates ring closing, and Rh-catalyzed hydrogenation of the unsaturated cyclic peptide results in selective formation of the natural product or its epimer, depending on our choice of phosphine ligand. NMR analysis and molecular modeling revealed that the linear peptide adopts a left-handed α-turn that preorganizes the N- and C-termini toward macrocyclization.
Co-reporter:Dr. Xuesong Wu;Jan Riedel; Vy M. Dong
Angewandte Chemie 2017 Volume 129(Issue 38) pp:11747-11751
Publication Date(Web):2017/09/11
DOI:10.1002/ange.201705859
AbstractWe have developed a strategy to transform olefins into homoallylic nitriles through a mechanism that combines copper catalysis with alkyl nitrile radicals. The radicals are easily generated from alkyl nitriles in the presence of the mild oxidant di-tert-butyl peroxide. This cross-dehydrogenative coupling between simple olefins and alkylnitriles bears advantages over the conventional use of halides and toxic cyanide reagents. With this method, we showcase the facile synthesis of a flavoring agent, a natural product, and a polymer precursor from simple olefins.
Co-reporter:Jung-Woo Park; Zhiwei Chen
Journal of the American Chemical Society 2016 Volume 138(Issue 10) pp:3310-3313
Publication Date(Web):March 8, 2016
DOI:10.1021/jacs.6b01445
We report a Rh-catalyzed enantioselective cycloisomerization of α,ω-heptadienes to afford cyclohexenes bearing quaternary carbon centers. Rhodium(I) and a new SDP ligand promote chemoselective formation of a cyclohex-3-enecarbaldehyde motif that is inaccessible by the Diels–Alder cycloaddition. Various α,α-bisallylaldehydes rearrange to generate six-membered rings by a mechanism triggered by aldehyde C–H bond activation. Mechanistic studies suggest a pathway involving regioselective carbometalation and endocyclic β-hydride elimination.
Co-reporter:Xuesong Wu, Zhiwei Chen, Yu-Bin Bai, and Vy M. Dong
Journal of the American Chemical Society 2016 Volume 138(Issue 37) pp:12013-12016
Publication Date(Web):September 1, 2016
DOI:10.1021/jacs.6b06227
We present a diastereodivergent strategy for constructing bicyclic γ-lactones bearing quaternary carbon centers via ketone hydroacylation. By applying a Rh catalyst and JoSPOphos ligand, either the anti or syn bicyclic γ-lactones can be accessed with high enantio- and diastereoselectivities, depending on the choice of solvent, temperature, and counterion.
Co-reporter:Faben A. Cruz, Zhiwei Chen, Sarah I. Kurtoic and Vy M. Dong
Chemical Communications 2016 vol. 52(Issue 34) pp:5836-5839
Publication Date(Web):29 Mar 2016
DOI:10.1039/C6CC02522F
Herein, we describe a regioselective Rh-catalyzed decarboxylative cross-coupling of β-keto acids and alkynes to access branched γ,δ-unsaturated ketones. Rh-hydride catalysis enables the isomerization of an alkyne to generate a metal-allyl species that can undergo carbon–carbon bond formation. Ketones are generated under mild conditions, without the need for base or activated electrophiles.
Co-reporter:Qing-An Chen; Faben A. Cruz
Journal of the American Chemical Society 2015 Volume 137(Issue 9) pp:3157-3160
Publication Date(Web):January 21, 2015
DOI:10.1021/ja512015w
By using tandem Ru-catalysis, internal alkynes can be coupled with aldehydes for the synthesis of β,γ-unsaturated ketones. The catalyst promotes alkyne transformations with high regioselectivity, with examples that include the differentiation of a methyl vs ethyl substituent on the alkyne. Mechanistic studies suggest that the regioselectivity results from a selective allene formation that is governed by allylic strain.
Co-reporter:Qing-An Chen; Zhiwei Chen
Journal of the American Chemical Society 2015 Volume 137(Issue 26) pp:8392-8395
Publication Date(Web):June 24, 2015
DOI:10.1021/jacs.5b05200
The hydroamination of internal alkynes via tandem rhodium catalysis gives branched N-allylic indolines with high regio- and enantioselectivity. An acid switch provides access to the linear isomer in preference to the branched isomer by an isomerization mechanism. Mechanistic studies suggest formation of an allene intermediate, which undergoes hydroamination to generate allylic amines instead of the enamine or imine products typically observed in alkyne hydroaminations.
Co-reporter:Jung-Woo Park, Kevin G. M. Kou, Daniel K. Kim and Vy M. Dong
Chemical Science 2015 vol. 6(Issue 8) pp:4479-4483
Publication Date(Web):12 Jun 2015
DOI:10.1039/C5SC01553G
We describe a Rh-catalyzed desymmetrization of all-carbon quaternary centers from α,α-bis(allyl)aldehydes by a cascade featuring isomerization and hydroacylation. This desymmetrization competes with two other novel olefin functionalizations that are triggered by C–H bond activation, including carboacylation and bisacylation. A BIPHEP ligand promotes enantioselective formation of α-vinylcyclopentanones. Mechanistic studies support irreversible and enantioselective olefin-isomerization followed by olefin-hydroacylation.
Co-reporter:Stephen K. Murphy, Achim Bruch and Vy M. Dong
Chemical Science 2015 vol. 6(Issue 1) pp:174-180
Publication Date(Web):22 Sep 2014
DOI:10.1039/C4SC02026J
The combination of a small-bite-angle diphosphine bis(dicyclohexylphosphino)methane (dcpm) and [Rh(cod)OMe]2 catalyses the hydroacylation of 2-vinylphenols with a wide range of non-chelating aldehydes. Here we present a detailed experimental study that elucidates the factors contributing to the broad aldehyde scope and high reactivity. A variety of catalytically relevant intermediates were isolated and a [Rh(dcpm)(vinylphenolate)] complex was identified as the major catalytically relevant species. A variety of off-cycle intermediates were also identified that can re-enter the catalytic cycle by substrate- or 1,5-cyclooctadiene-mediated pathways. Saturation kinetics with respect to the 2-vinylphenol were observed, and this may contribute to the high selectivity for hydroacylation over aldehyde decarbonylation. A series of deuterium labelling experiments and Hammett studies support the oxidative addition of Rh to the aldehyde C–H bond as an irreversible and turnover-limiting step. The small bite angle of dcpm is crucial for lowering the barrier of this step and providing excellent reactivity with a variety of aldehydes.
Co-reporter:Kevin G. M. Kou;Lauren E. Longobardi
Advanced Synthesis & Catalysis 2015 Volume 357( Issue 10) pp:2233-2237
Publication Date(Web):
DOI:10.1002/adsc.201500313
Co-reporter:K. G. M. Kou and V. M. Dong
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 21) pp:5844-5847
Publication Date(Web):05 May 2015
DOI:10.1039/C5OB00083A
Sulfoxides are uncommon substrates for transition-metal catalysis due to their propensity to inhibit catalyst turnover. In a collaborative effort with Ken Houk, we developed the first dynamic kinetic resolution (DKR) of allylic sulfoxides using asymmetric rhodium-catalyzed hydrogenation. A detailed mechanistic analysis of this transformation using both experimental and theoretical methods revealed rhodium to be a tandem catalyst that promoted both hydrogenation of the alkene and racemization of the allylic sulfoxide. Using a combination of deuterium labelling and DFT studies, a novel mode of allylic sulfoxide racemization via a Rh(III)-π-allyl intermediate was identified.
Co-reporter:Dr. Aaron M. Whittaker ;Dr. Vy M. Dong
Angewandte Chemie International Edition 2015 Volume 54( Issue 4) pp:1312-1315
Publication Date(Web):
DOI:10.1002/anie.201410322
Abstract
By exploring a new mode of nickel-catalyzed cross-coupling, a method to directly transform both aromatic and aliphatic aldehydes into either esters or amides has been developed. The success of this oxidative coupling depends on the appropriate choice of catalyst and organic oxidant, including the use of either α,α,α-trifluoroacetophenone or excess aldehyde. Mechanistic data that supports a catalytic cycle involving oxidative addition into the aldehyde CH bond is also presented.
Co-reporter:Dr. Aaron M. Whittaker ;Dr. Vy M. Dong
Angewandte Chemie 2015 Volume 127( Issue 4) pp:1328-1331
Publication Date(Web):
DOI:10.1002/ange.201410322
Abstract
By exploring a new mode of nickel-catalyzed cross-coupling, a method to directly transform both aromatic and aliphatic aldehydes into either esters or amides has been developed. The success of this oxidative coupling depends on the appropriate choice of catalyst and organic oxidant, including the use of either α,α,α-trifluoroacetophenone or excess aldehyde. Mechanistic data that supports a catalytic cycle involving oxidative addition into the aldehyde CH bond is also presented.
Co-reporter:Stephen K. Murphy;Jung-Woo Park;Faben A. Cruz
Science 2015 Volume 347(Issue 6217) pp:56-60
Publication Date(Web):02 Jan 2015
DOI:10.1126/science.1261232
Shifting hydroformylation into reverse
The hydroformylation reaction is applied on large scale in the chemical industry to make aldehydes by adding hydrogen and carbon monoxide to olefins. The reverse process could also prove useful in modifying complex molecules for pharmaceutical research, but methods directed toward that end often strip off the CO without the hydrogen. Murphy et al. now show that a rhodium catalyst can achieve selective dehydroformylation of a diverse range of compounds under mild conditions (see the Perspective by Landis). The protocol relies on effective transfer of the CO and H2 equivalents to a sacrificial strained olefin added to the mix.
Science, this issue p. 56; see also p. 29
Co-reporter:Kevin G. M. Kou ; Diane N. Le
Journal of the American Chemical Society 2014 Volume 136(Issue 26) pp:9471-9476
Publication Date(Web):June 17, 2014
DOI:10.1021/ja504296x
Under Rh(I) catalysis, α-ketoamides undergo intermolecular hydroacylation with aliphatic aldehydes. A newly designed Josiphos ligand enables access to α-acyloxyamides with high atom-economy and enantioselectivity. On the basis of mechanistic and kinetic studies, we propose a pathway in which rhodium plays a dual role in activating the aldehyde for cross-coupling. A stereochemical model is provided to rationalize the sense of enantioinduction observed.
Co-reporter:Nathan J. Oldenhuis ; Vy M. Dong ;Zhibin Guan
Journal of the American Chemical Society 2014 Volume 136(Issue 36) pp:12548-12551
Publication Date(Web):August 29, 2014
DOI:10.1021/ja5058482
A commercially available ruthenium(II) PNP-type pincer catalyst (Ru-Macho) promotes the formation of α-chiral tert-butanesulfinylamines from racemic secondary alcohols and Ellman’s chiral tert-butanesulfinamide via a hydrogen borrowing strategy. The formation of α-chiral tert-butanesulfinylamines occurs in yields ranging from 31% to 89% with most examples giving >95:5 dr.
Co-reporter:Stephen K. Murphy and Vy M. Dong
Chemical Communications 2014 vol. 50(Issue 89) pp:13645-13649
Publication Date(Web):03 Oct 2014
DOI:10.1039/C4CC02276A
Over thirty years ago, James and Young reported the first enantioselective olefin hydroacylation by using rhodium catalysts. This viewpoint highlights the advances in this area, including 4-pentenal cyclisations, medium-ring syntheses, and intermolecular variants.
Co-reporter:Dr. I-Hon Chen;Kevin G. M. Kou;Diane N. Le;Colin M. Rathbun;Dr. Vy M. Dong
Chemistry - A European Journal 2014 Volume 20( Issue 17) pp:5013-5018
Publication Date(Web):
DOI:10.1002/chem.201400133
Abstract
We demonstrate copper(II)-catalyzed acylation and tosylation of monosaccharides. Various carbohydrate derivatives, including glucopyranosides and ribofuranosides, are obtained in high yields and regioselectivities. Using this versatile strategy, the site of acylation can be switched by choice of ligand. Preliminary mechanistic studies support nucleophilic addition of a copper–sugar complex to the acyl chloride to be turnover limiting.
Co-reporter:Stephen K. Murphy;Dr. Achim Bruch;Dr. Vy M. Dong
Angewandte Chemie 2014 Volume 126( Issue 9) pp:2487-2491
Publication Date(Web):
DOI:10.1002/ange.201309987
Abstract
We report a protocol for the hydroacylation of vinylphenols with aryl, alkenyl, and alkyl aldehydes to form branched products with high selectivity. This cross-coupling yields α-aryl ketones that can be cyclized to benzofurans, and it enables access to eupomatenoid natural products in four steps or less from eugenol. Excellent reactivity and high levels of regioselectivity for the formation of the branched products were observed. We propose that aldehyde decarbonylation is avoided by the use of an anionic directing group on the alkene and a diphosphine ligand with a small bite angle.
Co-reporter:Stephen K. Murphy;Dr. Achim Bruch;Dr. Vy M. Dong
Angewandte Chemie International Edition 2014 Volume 53( Issue 9) pp:2455-2459
Publication Date(Web):
DOI:10.1002/anie.201309987
Abstract
We report a protocol for the hydroacylation of vinylphenols with aryl, alkenyl, and alkyl aldehydes to form branched products with high selectivity. This cross-coupling yields α-aryl ketones that can be cyclized to benzofurans, and it enables access to eupomatenoid natural products in four steps or less from eugenol. Excellent reactivity and high levels of regioselectivity for the formation of the branched products were observed. We propose that aldehyde decarbonylation is avoided by the use of an anionic directing group on the alkene and a diphosphine ligand with a small bite angle.
Co-reporter:Nathan J. Oldenhuis, Vy M. Dong, Zhibin Guan
Tetrahedron 2014 70(27–28) pp: 4213-4218
Publication Date(Web):
DOI:10.1016/j.tet.2014.03.085
Co-reporter:Charles S. Yeung
Topics in Catalysis 2014 Volume 57( Issue 17-20) pp:1342-1350
Publication Date(Web):2014 November
DOI:10.1007/s11244-014-0301-9
Developing more green and sustainable strategies for organic synthesis is an important modern challenge. Although carbon dioxide is an attractive reagent, because of its availability, the low reactivity of this small molecule renders it challenging to use widely in organic synthesis. The discovery of new catalysts thus plays a central role in advancing the science and impact of CO2 utilization. This perspective describes advances in metal-catalyzed carbon dioxide incorporation since Aresta’s report of the first metal-CO2 complex.
Co-reporter:Peter K. Dornan ; Kevin G. M. Kou ; K. N. Houk
Journal of the American Chemical Society 2013 Volume 136(Issue 1) pp:291-298
Publication Date(Web):December 18, 2013
DOI:10.1021/ja409824b
A dynamic kinetic resolution (DKR) of allylic sulfoxides has been demonstrated by combining the Mislow [2,3]-sigmatropic rearrangement with catalytic asymmetric hydrogenation. The efficiency of our DKR was optimized by using low pressures of hydrogen gas to decrease the rate of hydrogenation relative to the rate of sigmatropic rearrangement. Kinetic studies reveal that the rhodium complex acts as a dual-role catalyst and accelerates the substrate racemization while catalyzing olefin hydrogenation. Scrambling experiments and theoretical modeling support a novel mode of sulfoxide racemization which occurs via a rhodium π-allyl intermediate in polar solvents. In nonpolar solvents, however, the substrate racemization is primarily uncatalyzed. Computational studies suggest that the sulfoxide binds to rhodium via O-coordination throughout the catalytic cycle for hydrogenation.
Co-reporter:Stephen K. Murphy
Journal of the American Chemical Society 2013 Volume 135(Issue 15) pp:5553-5556
Publication Date(Web):April 8, 2013
DOI:10.1021/ja4021974
An enantioselective ketone hydroacylation enables the direct preparation of lactones from keto alcohols. The alcohol is oxidized in situ to an aldehyde, obviating the need to prepare sensitive keto aldehyde substrates. Noyori’s asymmetric transfer hydrogenation catalyst was applied to address challenges of reactivity, chemoselectivity, and enantioselectivity.
Co-reporter:Diem T.H. Phan, Vy M. Dong
Tetrahedron 2013 69(27–28) pp: 5726-5731
Publication Date(Web):
DOI:10.1016/j.tet.2013.04.133
Co-reporter:Max von Delius ; Christine M. Le
Journal of the American Chemical Society () pp:
Publication Date(Web):August 31, 2012
DOI:10.1021/ja305593y
We describe a method that allows salicylaldehyde derivatives to be coupled with a wide range of unactivated alkenes at catalyst loadings as low as 2 mol %. A chiral phosphoramidite ligand and the precise stoichiometry of heterogeneous base are key for high catalytic activity and linear regioselectivity. This protocol was applied in the atom- and step-economical synthesis of eight biologically active octaketide natural products, including anticancer drug candidate cytosporone B. Mechanistic studies provide insight on parameters affecting decarbonylation, a side reaction that limits the turnover number for catalytic hydroacylation. Deuterium labeling studies show that branched hydride insertion is fully reversible, whereas linear hydride insertion is largely irreversible and turnover-limiting. We propose that ligand (Ra,R,R)-SIPHOS-PE effectively suppresses decarbonylation, and helps favor a turnover-limiting insertion, by lowering the barrier for reductive elimination in the linear-selective pathway. Together, these factors enable high reactivity and regioselectivity.
Co-reporter:Faben A. Cruz, Zhiwei Chen, Sarah I. Kurtoic and Vy M. Dong
Chemical Communications 2016 - vol. 52(Issue 34) pp:NaN5839-5839
Publication Date(Web):2016/03/29
DOI:10.1039/C6CC02522F
Herein, we describe a regioselective Rh-catalyzed decarboxylative cross-coupling of β-keto acids and alkynes to access branched γ,δ-unsaturated ketones. Rh-hydride catalysis enables the isomerization of an alkyne to generate a metal-allyl species that can undergo carbon–carbon bond formation. Ketones are generated under mild conditions, without the need for base or activated electrophiles.
Co-reporter:Stephen K. Murphy, Achim Bruch and Vy M. Dong
Chemical Science (2010-Present) 2015 - vol. 6(Issue 1) pp:NaN180-180
Publication Date(Web):2014/09/22
DOI:10.1039/C4SC02026J
The combination of a small-bite-angle diphosphine bis(dicyclohexylphosphino)methane (dcpm) and [Rh(cod)OMe]2 catalyses the hydroacylation of 2-vinylphenols with a wide range of non-chelating aldehydes. Here we present a detailed experimental study that elucidates the factors contributing to the broad aldehyde scope and high reactivity. A variety of catalytically relevant intermediates were isolated and a [Rh(dcpm)(vinylphenolate)] complex was identified as the major catalytically relevant species. A variety of off-cycle intermediates were also identified that can re-enter the catalytic cycle by substrate- or 1,5-cyclooctadiene-mediated pathways. Saturation kinetics with respect to the 2-vinylphenol were observed, and this may contribute to the high selectivity for hydroacylation over aldehyde decarbonylation. A series of deuterium labelling experiments and Hammett studies support the oxidative addition of Rh to the aldehyde C–H bond as an irreversible and turnover-limiting step. The small bite angle of dcpm is crucial for lowering the barrier of this step and providing excellent reactivity with a variety of aldehydes.
Co-reporter:K. G. M. Kou and V. M. Dong
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 21) pp:NaN5847-5847
Publication Date(Web):2015/05/05
DOI:10.1039/C5OB00083A
Sulfoxides are uncommon substrates for transition-metal catalysis due to their propensity to inhibit catalyst turnover. In a collaborative effort with Ken Houk, we developed the first dynamic kinetic resolution (DKR) of allylic sulfoxides using asymmetric rhodium-catalyzed hydrogenation. A detailed mechanistic analysis of this transformation using both experimental and theoretical methods revealed rhodium to be a tandem catalyst that promoted both hydrogenation of the alkene and racemization of the allylic sulfoxide. Using a combination of deuterium labelling and DFT studies, a novel mode of allylic sulfoxide racemization via a Rh(III)-π-allyl intermediate was identified.
Co-reporter:Jung-Woo Park, Kevin G. M. Kou, Daniel K. Kim and Vy M. Dong
Chemical Science (2010-Present) 2015 - vol. 6(Issue 8) pp:NaN4483-4483
Publication Date(Web):2015/06/12
DOI:10.1039/C5SC01553G
We describe a Rh-catalyzed desymmetrization of all-carbon quaternary centers from α,α-bis(allyl)aldehydes by a cascade featuring isomerization and hydroacylation. This desymmetrization competes with two other novel olefin functionalizations that are triggered by C–H bond activation, including carboacylation and bisacylation. A BIPHEP ligand promotes enantioselective formation of α-vinylcyclopentanones. Mechanistic studies support irreversible and enantioselective olefin-isomerization followed by olefin-hydroacylation.
Co-reporter:Stephen K. Murphy and Vy M. Dong
Chemical Communications 2014 - vol. 50(Issue 89) pp:NaN13649-13649
Publication Date(Web):2014/10/03
DOI:10.1039/C4CC02276A
Over thirty years ago, James and Young reported the first enantioselective olefin hydroacylation by using rhodium catalysts. This viewpoint highlights the advances in this area, including 4-pentenal cyclisations, medium-ring syntheses, and intermolecular variants.
.jpg)
![(Z)-TERT-BUTYL-[2-[(1S)-1-DIPHENYLPHOSPHANYLETHYL]CYCLOPENTA-2,4-DIEN-1-YLIDENE]-OXIDOPHOSPHANIUM;CYCLOPENTA-1,3-DIENE;IRON(2+)](http://img.cochemist.com/ccimg/1221800/1221746-31-1.png)
![(Z)-TERT-BUTYL-[2-[(1S)-1-DIPHENYLPHOSPHANYLETHYL]CYCLOPENTA-2,4-DIEN-1-YLIDENE]-OXIDOPHOSPHANIUM;CYCLOPENTA-1,3-DIENE;IRON(2+)](http://img.cochemist.com/ccimg/1221800/1221746-31-1_b.png)
![(Z)-TERT-BUTYL-[2-[(1R)-1-DIPHENYLPHOSPHANYLETHYL]CYCLOPENTA-2,4-DIEN-1-YLIDENE]-OXIDOPHOSPHANIUM;CYCLOPENTA-1,3-DIENE;IRON(2+)](http://img.cochemist.com/ccimg/1221800/1221745-90-9.png)
![(Z)-TERT-BUTYL-[2-[(1R)-1-DIPHENYLPHOSPHANYLETHYL]CYCLOPENTA-2,4-DIEN-1-YLIDENE]-OXIDOPHOSPHANIUM;CYCLOPENTA-1,3-DIENE;IRON(2+)](http://img.cochemist.com/ccimg/1221800/1221745-90-9_b.png)
![2,2',3,3'-Tetrahydro-1,1'-spirobi[indene]-7,7'-diylbis[bis(3,5-di methylphenyl)phosphine]](/data/chemimg/2243100/917377-75-4.png)
![2,2',3,3'-Tetrahydro-1,1'-spirobi[indene]-7,7'-diylbis[bis(3,5-di methylphenyl)phosphine]](/data/chemimg/2243100/917377-75-4_b.png)
![Phosphine,1,1'-[(1R)-2,2',3,3'-tetrahydro-1,1'-spirobi[1H-indene]-7,7'-diyl]bis[1,1-diphenyl-(9CI)](/data/chemimg/1006900/917377-74-3.png)
![Phosphine,1,1'-[(1R)-2,2',3,3'-tetrahydro-1,1'-spirobi[1H-indene]-7,7'-diyl]bis[1,1-diphenyl-(9CI)](/data/chemimg/1006900/917377-74-3_b.png)



![1,3-CYCLOHEXANEDIONE, 2-[(4-FLUOROPHENYL)METHYL]-](http://img.cochemist.com/ccimg/724500/724456-36-4.png)
![1,3-CYCLOHEXANEDIONE, 2-[(4-FLUOROPHENYL)METHYL]-](http://img.cochemist.com/ccimg/724500/724456-36-4_b.png)


![Phosphine oxide, bis[3,5-bis(1,1-dimethylethyl)-4-methoxyphenyl]-](http://img.cochemist.com/ccimg/536000/535925-40-7.png)
![Phosphine oxide, bis[3,5-bis(1,1-dimethylethyl)-4-methoxyphenyl]-](http://img.cochemist.com/ccimg/536000/535925-40-7_b.png)



![Benzenamine, N,N-bis[(2E)-3-phenyl-2-propenyl]-](http://img.cochemist.com/ccimg/234100/234092-61-6.png)
![Benzenamine, N,N-bis[(2E)-3-phenyl-2-propenyl]-](http://img.cochemist.com/ccimg/234100/234092-61-6_b.png)



![Benzenemethanamine, N-(phenylmethyl)-N-[(2E)-3-phenyl-2-propen-1-yl]-](/data/chemimg/109900/147452-43-5.png)
![Benzenemethanamine, N-(phenylmethyl)-N-[(2E)-3-phenyl-2-propen-1-yl]-](/data/chemimg/109900/147452-43-5_b.png)


















![Carbonochloridic acid,(1S,2R,4S)-1,7,7-trimethylbicyclo[2.2.1]hept-2-yl ester](http://img.cochemist.com/ccimg/104600/104528-61-2.png)
![Carbonochloridic acid,(1S,2R,4S)-1,7,7-trimethylbicyclo[2.2.1]hept-2-yl ester](http://img.cochemist.com/ccimg/104600/104528-61-2_b.png)
![1H-Indole, 3-acetyl-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/104200/104142-24-7.png)
![1H-Indole, 3-acetyl-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/104200/104142-24-7_b.png)
![1H-INDOLE, 5-BROMO-1-[(4-METHYLPHENYL)SULFONYL]-](http://img.cochemist.com/ccimg/96600/96546-77-9.png)
![1H-INDOLE, 5-BROMO-1-[(4-METHYLPHENYL)SULFONYL]-](http://img.cochemist.com/ccimg/96600/96546-77-9_b.png)
![Benzenamine, N-[(2E)-3-phenyl-2-propenyl]-](http://img.cochemist.com/ccimg/92600/92573-86-9.png)
![Benzenamine, N-[(2E)-3-phenyl-2-propenyl]-](http://img.cochemist.com/ccimg/92600/92573-86-9_b.png)




















![L-TYROSINE, N-[N-[(1,1-DIMETHYLETHOXY)CARBONYL]GLYCYL]-, METHYL ESTER](http://img.cochemist.com/ccimg/65200/65160-62-5.png)
![L-TYROSINE, N-[N-[(1,1-DIMETHYLETHOXY)CARBONYL]GLYCYL]-, METHYL ESTER](http://img.cochemist.com/ccimg/65200/65160-62-5_b.png)
![PYRROLIDINE, 2-(IODOMETHYL)-1-[(4-METHYLPHENYL)SULFONYL]-, (S)-](http://img.cochemist.com/ccimg/63400/63328-01-8.png)
![PYRROLIDINE, 2-(IODOMETHYL)-1-[(4-METHYLPHENYL)SULFONYL]-, (S)-](http://img.cochemist.com/ccimg/63400/63328-01-8_b.png)




![[1,1'-Biphenyl]-4-aceticacid, methyl ester](http://img.cochemist.com/ccimg/59800/59793-29-2.png)
![[1,1'-Biphenyl]-4-aceticacid, methyl ester](http://img.cochemist.com/ccimg/59800/59793-29-2_b.png)












































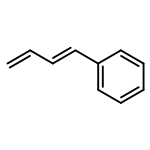
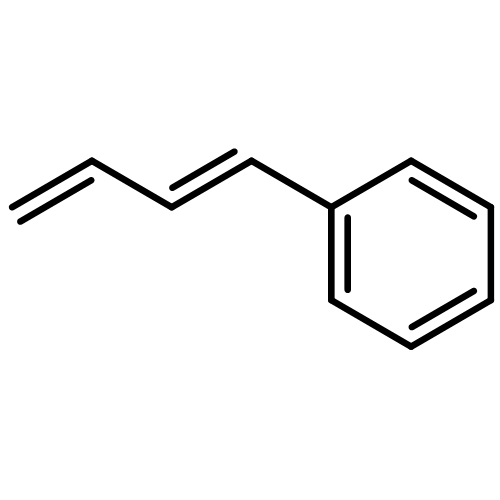




![bis[(2,3,5,6-η)-bicyclo[2.2.1]hepta-2,5-diene]di-μ-chlorodirhodium](http://img.cochemist.com/ccimg/12300/12257-42-0.png)
![bis[(2,3,5,6-η)-bicyclo[2.2.1]hepta-2,5-diene]di-μ-chlorodirhodium](http://img.cochemist.com/ccimg/12300/12257-42-0_b.png)
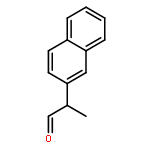
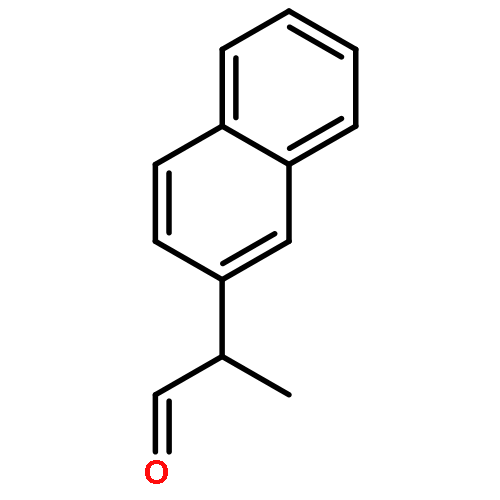





![N-[2-(hydroxymethyl)phenyl]benzamide](http://img.cochemist.com/ccimg/6300/6289-87-8.png)
![N-[2-(hydroxymethyl)phenyl]benzamide](http://img.cochemist.com/ccimg/6300/6289-87-8_b.png)






![Benzene, [(2-butynyloxy)methyl]-](http://img.cochemist.com/ccimg/4900/4885-07-8.png)
![Benzene, [(2-butynyloxy)methyl]-](http://img.cochemist.com/ccimg/4900/4885-07-8_b.png)















![L-Phenylalanine, N-[N-[(1,1-dimethylethoxy)carbonyl]-L-alanyl]-](http://img.cochemist.com/ccimg/2500/2448-58-0.png)
![L-Phenylalanine, N-[N-[(1,1-dimethylethoxy)carbonyl]-L-alanyl]-](http://img.cochemist.com/ccimg/2500/2448-58-0_b.png)
![L-Phenylalanine, N-[(1,1-dimethylethoxy)carbonyl]-L-alanyl-, methyl ester](http://img.cochemist.com/ccimg/2300/2280-66-2.png)
![L-Phenylalanine, N-[(1,1-dimethylethoxy)carbonyl]-L-alanyl-, methyl ester](http://img.cochemist.com/ccimg/2300/2280-66-2_b.png)








![(8R,9S,13S,14S)-13-methyl-6,7,8,9,11,12,13,14,15,16-decahydrospiro[cyclopenta[a]phenanthrene-17,2'-[1,3]dioxolan]-3-ol](http://img.cochemist.com/ccimg/1000/900-83-4.png)
![(8R,9S,13S,14S)-13-methyl-6,7,8,9,11,12,13,14,15,16-decahydrospiro[cyclopenta[a]phenanthrene-17,2'-[1,3]dioxolan]-3-ol](http://img.cochemist.com/ccimg/1000/900-83-4_b.png)






















![Rhodium, bis[(1,2,5,6-h)-1,5-cyclooctadiene]di-m-methoxydi-](http://img.cochemist.com/ccimg/12200/12148-72-0.png)
![Rhodium, bis[(1,2,5,6-h)-1,5-cyclooctadiene]di-m-methoxydi-](http://img.cochemist.com/ccimg/12200/12148-72-0_b.png)