Co-reporter:Lizbeth Hedstrom
ACS Infectious Diseases January 13, 2017 Volume 3(Issue 1) pp:
Publication Date(Web):November 9, 2016
DOI:10.1021/acsinfecdis.6b00185
The convergence of competitive fitness experiments and phenotypic screening would seem to be an auspicious beginning for validation of an antibacterial target. IMPDH was already identified an essential protein in Mycobacterium tuberculosis when not one, but two, groups discovered inhibitors with promising antitubercular activity. A new target appeared to be born. Surprisingly, the two groups came to completely different conclusions about the vulnerability of IMPDH and its future as a drug target. This viewpoint discusses these papers and how to resolve this conundrum.
Co-reporter:Yang Wei, Petr Kuzmič, Runhan Yu, Gyan Modi, and Lizbeth Hedstrom
Biochemistry 2016 Volume 55(Issue 37) pp:5279
Publication Date(Web):August 19, 2016
DOI:10.1021/acs.biochem.6b00265
Inosine-5′-monophosphate dehydrogenase (IMPDH) catalyzes the conversion of inosine 5′-monophosphate (IMP) to xanthosine 5′-monophosphate (XMP). The enzyme is an emerging target for antimicrobial therapy. The small molecule inhibitor A110 has been identified as a potent and selective inhibitor of IMPDHs from a variety of pathogenic microorganisms. A recent X-ray crystallographic study reported that the inhibitor binds to the NAD+ cofactor site and forms a ternary complex with IMP. Here we report a pre-steady-state stopped-flow kinetic investigation of IMPDH from Bacillus anthracis designed to assess the kinetic significance of the crystallographic results. Stopped-flow kinetic experiments defined nine microscopic rate constants and two equilibrium constants that characterize both the catalytic cycle and details of the inhibition mechanism. In combination with steady-state initial rate studies, the results show that the inhibitor binds with high affinity (Kd ≈ 50 nM) predominantly to the covalent intermediate on the reaction pathway. Only a weak binding interaction (Kd ≈ 1 μM) is observed between the inhibitor and E·IMP. Thus, the E·IMP·A110 ternary complex, observed by X-ray crystallography, is largely kinetically irrelevant.
Co-reporter:Yuntao Shi, Marcus J.C. Long, Masha M. Rosenberg, Shican Li, Aimee Kobjack, Philip Lessans, Rory T. Coffey, and Lizbeth Hedstrom
ACS Chemical Biology 2016 Volume 11(Issue 12) pp:
Publication Date(Web):October 5, 2016
DOI:10.1021/acschembio.6b00656
Targeted protein degradation is a promising strategy for drug design and functional assessment. Several small molecule approaches have been developed that localize target proteins to ubiquitin ligases, inducing ubiquitination and subsequent degradation by the 26S proteasome. We discovered that the degradation of a target protein can also be induced by a recognition ligand linked to tert-butyl carbamate (Boc3)-protected arginine (B3A). Here, we show that this process requires the proteasome but does not involve ubiquitination of the target protein. B3A does not perturb the structure of the target protein; instead, a B3A-ligand stabilizes its target protein. B3A ligands stimulate activity of purified 20S proteasome, demonstrating that the tag binds directly to the 20S proteasome. Moreover, purified 20S proteasome is sufficient to degrade target proteins in the presence of their respective B3A-linked recognition ligands. These observations suggest a simple model for B3A-mediated degradation wherein the B3A tag localizes target proteins directly to the 20S proteasome. Thus, B3A ligands are the first example of a ubiquitin-free strategy for targeted protein degradation.
Co-reporter:Youngchang Kim;Magdalena Makowska-Grzyska;Suresh Kumar Gorla;Deviprasad R. Gollapalli;Gregory D. Cuny;Andrzej Joachimiak
Acta Crystallographica Section F 2015 Volume 71( Issue 5) pp:531-538
Publication Date(Web):
DOI:10.1107/S2053230X15000187
Inosine 5′-monophosphate dehydrogenase (IMPDH) is a promising target for the treatment of Cryptosporidium infections. Here, the structure of C. parvum IMPDH (CpIMPDH) in complex with inosine 5′-monophosphate (IMP) and P131, an inhibitor with in vivo anticryptosporidial activity, is reported. P131 contains two aromatic groups, one of which interacts with the hypoxanthine ring of IMP, while the second interacts with the aromatic ring of a tyrosine in the adjacent subunit. In addition, the amine and NO2 moieties bind in hydrated cavities, forming water-mediated hydrogen bonds to the protein. The design of compounds to replace these water molecules is a new strategy for the further optimization of C. parvum inhibitors for both antiparasitic and antibacterial applications.
Co-reporter:Kavitha Mandapati, Suresh Kumar Gorla, Amanda L. House, Elizabeth S. McKenney, Minjia Zhang, Suraj Nagendra Rao, Deviprasad R. Gollapalli, Barbara J. Mann, Joanna B. Goldberg, Gregory D. Cuny, Ian J. Glomski, and Lizbeth Hedstrom
ACS Medicinal Chemistry Letters 2014 Volume 5(Issue 8) pp:846
Publication Date(Web):June 10, 2014
DOI:10.1021/ml500203p
Inosine 5′-monophosphate dehydrogenase (IMPDH) catalyzes the pivotal step in guanine nucleotide biosynthesis. IMPDH is a target for immunosuppressive, antiviral, and anticancer drugs, but, as of yet, has not been exploited for antimicrobial therapy. We have previously reported potent inhibitors of IMPDH from the protozoan parasite Cryptosporidium parvum (CpIMPDH). Many pathogenic bacteria, including Bacillus anthracis, Staphylococcus aureus, and Listeria monocytogenes, contain IMPDHs that should also be inhibited by these compounds. Herein, we present the structure–activity relationships for the inhibition of B. anthracis IMPDH (BaIMPDH) and antibacterial activity of 140 compounds from five structurally distinct compound series. Many potent inhibitors of BaIMPDH were identified (78% with IC50 ≤ 1 μM). Four compounds had minimum inhibitory concentrations (MIC) of less than 2 μM against B. anthracis Sterne 770. These compounds also displayed antibacterial activity against S. aureus and L. monocytogenes.Keywords: antibacterial; antibiotic; Gram-positive; IMP dehydrogenase; inhibitor
Co-reporter:Corey R. Johnson, Suresh Kumar Gorla, Mandapati Kavitha, Minjia Zhang, Xiaoping Liu, Boris Striepen, Jan R. Mead, Gregory D. Cuny, Lizbeth Hedstrom
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 4) pp:1004-1007
Publication Date(Web):15 February 2013
DOI:10.1016/j.bmcl.2012.12.037
Cryptosporidium parvum (Cp) is a potential biowarfare agent and major cause of diarrhea and malnutrition. This protozoan parasite relies on inosine 5′-monophosphate dehydrogenase (IMPDH) for the production of guanine nucleotides. A CpIMPDH-selective N-aryl-3,4-dihydro-3-methyl-4-oxo-1-phthalazineacetamide inhibitor was previously identified in a high throughput screening campaign. Herein we report a structure–activity relationship study for the phthalazinone-based series that resulted in the discovery of benzofuranamide analogs that exhibit low nanomolar inhibition of CpIMPDH. In addition, the antiparasitic activity of select analogs in a Toxoplasma gondii model of C. parvum infection is also presented.
Co-reporter:Marcus J.C. Long, Deviprasad R. Gollapalli, Lizbeth Hedstrom
Chemistry & Biology 2012 Volume 19(Issue 5) pp:629-637
Publication Date(Web):25 May 2012
DOI:10.1016/j.chembiol.2012.04.008
The discovery of drugs that cause the degradation of their target proteins has been largely serendipitous. Here we report that the tert-butyl carbamate-protected arginine (Boc3Arg) moiety provides a general strategy for the design of degradation-inducing inhibitors. The covalent inactivators ethacrynic acid and thiobenzofurazan cause the specific degradation of glutathione-S-transferase when linked to Boc3Arg. Similarly, the degradation of dihydrofolate reductase is induced when cells are treated with the noncovalent inhibitor trimethoprim linked to Boc3Arg. Degradation is rapid and robust, with 30%–80% of these abundant target proteins consumed within 1.3–5 hr. The proteasome is required for Boc3Arg-mediated degradation, but ATP is not necessary and the ubiquitin pathways do not appear to be involved. These results suggest that the Boc3Arg moiety may provide a general strategy to construct inhibitors that induce targeted protein degradation.Graphical AbstractFigure optionsDownload full-size imageDownload high-quality image (182 K)Download as PowerPoint slideHighlights► Two Boc3Arg-linked covalent inhibitors induce degradation of GST proteins in cells ► A Boc3Arg-linked noncovalent inhibitor induces degradation of DHFR in cells ► 30%–80% degradation of the target protein is observed within 1.3–5 hr ► Proteasome inhibitors block targeted degradation, but ubiquitylation is not required
Co-reporter:Sivapriya Kirubakaran, Suresh Kumar Gorla, Lisa Sharling, Minjia Zhang, Xiaoping Liu, Soumya S. Ray, Iain S. MacPherson, Boris Striepen, Lizbeth Hedstrom, Gregory D. Cuny
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 5) pp:1985-1988
Publication Date(Web):1 March 2012
DOI:10.1016/j.bmcl.2012.01.029
Cryptosporidium parasites are important waterborne pathogens of both humans and animals. The Cryptosporidium parvum and Cryptosporidium hominis genomes indicate that the only route to guanine nucleotides is via inosine 5′-monophosphate dehydrogenase (IMPDH). Thus the inhibition of the parasite IMPDH presents a potential strategy for treating Cryptosporidium infections. A selective benzimidazole-based inhibitor of C. parvum IMPDH (CpIMPDH) was previously identified in a high throughput screen. Here we report a structure–activity relationship study of benzimidazole-based compounds that resulted in potent and selective inhibitors of CpIMPDH. Several compounds display potent antiparasitic activity in vitro.
Co-reporter:Marcus J. C. Long; Lizbeth Hedstrom
ChemBioChem 2012 Volume 13( Issue 12) pp:1818-1825
Publication Date(Web):
DOI:10.1002/cbic.201100792
Abstract
We show that mushroom tyrosinase catalyzes the formation of reactive o-quinones on unstructured, tyrosine-rich sequences such as hemagglutinin (HA) tags (YPYDVPDYA). In the absence of exogenous nucleophiles and at low protein concentrations, the o-quinone decomposes with fragmentation of the HA tag. At higher protein concentrations (>5 mg mL−1), crosslinking is observed. Besthorn's reagent intercepts the o-quinone to give a characteristic pink complex that can be observed directly on a denaturing SDS-PAGE gel. Similar labeled species can be formed by using other nucleophiles such as Cy5-hydrazide. These reactions are selective for proteins bearing HA and other unstructured poly-tyrosine-containing tags and can be performed in lysates to create specifically tagged proteins.
Co-reporter:Thomas V. Riera, Lianqing Zheng, Helen R. Josephine, Donghong Min, Wei Yang, and Lizbeth Hedstrom
Biochemistry 2011 Volume 50(Issue 39) pp:
Publication Date(Web):August 26, 2011
DOI:10.1021/bi200785s
Allosteric activators are generally believed to shift the equilibrium distribution of enzyme conformations to favor a catalytically productive structure; the kinetics of conformational exchange is seldom addressed. Several observations suggested that the usual allosteric mechanism might not apply to the activation of IMP dehydrogenase (IMPDH) by monovalent cations. Therefore, we investigated the mechanism of K+ activation in IMPDH by delineating the kinetic mechanism in the absence of monovalent cations. Surprisingly, the K+ dependence of kcat derives from the rate of flap closure, which increases by ≥65-fold in the presence of K+. We performed both alchemical free energy simulations and potential of mean force calculations using the orthogonal space random walk strategy to computationally analyze how K+ accelerates this conformational change. The simulations recapitulate the preference of IMPDH for K+, validating the computational models. When K+ is replaced with a dummy ion, the residues of the K+ binding site relax into ordered secondary structure, creating a barrier to conformational exchange. K+ mobilizes these residues by providing alternate interactions for the main chain carbonyls. Potential of mean force calculations indicate that K+ changes the shape of the energy well, shrinking the reaction coordinate by shifting the closed conformation toward the open state. This work suggests that allosteric regulation can be under kinetic as well as thermodynamic control.
Co-reporter:B. Christopher Hoefler, Deviprasad R. Gollapalli, Lizbeth Hedstrom
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 5) pp:1363-1365
Publication Date(Web):1 March 2011
DOI:10.1016/j.bmcl.2011.01.042
IMP dehydrogenase (IMPDH) catalyzes a critical step in guanine nucleotide biosynthesis. IMPDH also has biological roles that are distinct from its enzymatic function. We report a biotin-linked reagent that selectively labels IMPDH and is released by dithiothreitol. This reagent will be invaluable in elucidating the moonlighting functions of IMPDH.
Co-reporter:Iain S. MacPherson ; Sivapriya Kirubakaran ; Suresh Kumar Gorla ; Thomas V. Riera ; J. Alejandro D’Aquino ; Minjia Zhang ; Gregory D. Cuny
Journal of the American Chemical Society 2010 Volume 132(Issue 4) pp:1230-1231
Publication Date(Web):January 6, 2010
DOI:10.1021/ja909947a
Cryptosporidium parvum is a potential biowarfare agent, an important AIDS pathogen, and a major cause of diarrhea and malnutrition. No vaccines or effective drug treatment exist to combat Cryptosporidium infection. This parasite relies on inosine 5′-monophosphate dehydrogenase (IMPDH) to obtain guanine nucleotides, and inhibition of this enzyme blocks parasite proliferation. Here, we report the first crystal structures of CpIMPDH. These structures reveal the structural basis of inhibitor selectivity and suggest a strategy for further optimization. Using this information, we have synthesized low-nanomolar inhibitors that display 103 selectivity for the parasite enzyme over human IMPDH2.
Co-reporter:Deviprasad R. Gollapalli, Iain S. MacPherson, George Liechti, Suresh Kumar Gorla, Joanna B. Goldberg, Lizbeth Hedstrom
Chemistry & Biology 2010 Volume 17(Issue 10) pp:1084-1091
Publication Date(Web):29 October 2010
DOI:10.1016/j.chembiol.2010.07.014
The protozoan parasite Cryptosporidium parvum is a major cause of gastrointestinal disease; no effective drug therapy exists to treat this infection. Curiously, C. parvum IMPDH (CpIMPDH) is most closely related to prokaryotic IMPDHs, suggesting that the parasite obtained its IMPDH gene via horizontal transfer. We previously identified inhibitors of CpIMPDH that do not inhibit human IMPDHs. Here, we show that these compounds also inhibit IMPDHs from Helicobacter pylori, Borrelia burgdorferi, and Streptococcus pyogenes, but not from Escherichia coli. Residues Ala165 and Tyr358 comprise a structural motif that defines susceptible enzymes. Importantly, a second-generation CpIMPDH inhibitor has bacteriocidal activity on H. pylori but not E. coli. We propose that CpIMPDH-targeted inhibitors can be developed into a new class of antibiotics that will spare some commensal bacteria.Highlights► CpIMPDH inhibitors also inhibit IMPDHs from H. pylori, B. burgdorferi, S. pyogenes, but not E. coli ► A second-generation CpIMPDH inhibitor blocks H. pylori but not E. coli growth ► The presence of Ala165 and Tyr358 comprise a structural motif that defines susceptible enzymes ► This motif is found in IMPDHs from a wide variety of pathogenic bacteria
Co-reporter:Helen R. Josephine, Kanchana R. Ravichandran, and Lizbeth Hedstrom
Biochemistry 2010 Volume 49(Issue 50) pp:
Publication Date(Web):November 9, 2010
DOI:10.1021/bi101590c
X-ray crystal structures of enzyme−ligand complexes are widely believed to mimic states in the catalytic cycle, but this presumption has seldom been carefully scrutinized. In the case of Tritrichomonas foetus inosine 5′-monophosphate dehydrogenase (IMPDH), 10 structures of various enzyme−substrate−inhibitor complexes have been determined. The Cys319 loop is found in at least three different conformations, suggesting that its conformation changes as the catalytic cycle progresses from the dehydrogenase step to the hydrolase reaction. Alternatively, only one conformation of the Cys319 loop may be catalytically relevant while the others are off-pathway. Here we differentiate between these two hypotheses by analyzing the effects of Ala substitutions at three residues of the Cys319 loop, Arg322, Glu323, and Gln324. These mutations have minimal effects on the value of kcat (≤5-fold) that obscure large effects (>10-fold) on the microscopic rate constants for individual steps. These substitutions increase the equilibrium constant for the dehydrogenase step but decrease the equilibrium between open and closed conformations of a mobile flap. More dramatic effects are observed when Arg322 is substituted with Glu, which decreases the rates of hydride transfer and hydrolysis by factors of 2000 and 130, respectively. These experiments suggest that the Cys319 loop does indeed have different conformations during the dehydrogenase and hydrolase reactions as suggested by the crystal structures. Importantly, these experiments reveal that the structure of the Cys319 loop modulates the closure of the mobile flap. This conformational change converts the enzyme from a dehydrogenase into hydrolase, suggesting that the conformation of the Cys319 loop may gate the catalytic cycle.
Co-reporter:Lizbeth Hedstrom
Chemical Reviews 2009 Volume 109(Issue 7) pp:2903
Publication Date(Web):May 29, 2009
DOI:10.1021/cr900021w
Co-reporter:Nwakaso N. Umejiego, Deviprasad Gollapalli, Lisa Sharling, Anna Volftsun, Jennifer Lu, Nicole N. Benjamin, Adam H. Stroupe, Thomas V. Riera, Boris Striepen, Lizbeth Hedstrom
Chemistry & Biology 2008 Volume 15(Issue 1) pp:70-77
Publication Date(Web):25 January 2008
DOI:10.1016/j.chembiol.2007.12.010
Cryptosporidium parvum is an important human pathogen and potential bioterrorism agent. No vaccines exist against C. parvum, the drugs currently approved to treat cryptosporidiosis are ineffective, and drug discovery is challenging because the parasite cannot be maintained continuously in cell culture. Mining the sequence of the C. parvum genome has revealed that the only route to guanine nucleotides is via inosine-5′-monophosphate dehydrogenase (IMPDH). Moreover, phylogenetic analysis suggests that the IMPDH gene was obtained from bacteria by lateral gene transfer. Here we exploit the unexpected evolutionary divergence of parasite and host enzymes by designing a high-throughput screen to target the most diverged portion of the IMPDH active site. We have identified four parasite-selective IMPDH inhibitors that display antiparasitic activity with greater potency than paromomycin, the current gold standard for anticryptosporidial activity.
Co-reporter:Thomas V. Riera, Wen Wang, Helen R. Josephine and Lizbeth Hedstrom
Biochemistry 2008 Volume 47(Issue 33) pp:
Publication Date(Web):July 22, 2008
DOI:10.1021/bi800674a
IMP dehydrogenase (IMPDH) catalyzes two very different chemical transformations, a dehydrogenase reaction and a hydrolysis reaction. The enzyme toggles between the open conformation required for the dehydrogenase reaction and the closed conformation of the hydrolase reaction by moving a mobile flap into the NAD site. Despite these multiple functional constraints, the residues of the flap and NAD site are highly diverged, and the equilibrium between open and closed conformations (Kc) varies widely. In order to understand how differences in the dynamic properties of the flap influence the catalytic cycle, we have delineated the kinetic mechanism of IMPDH from the pathogenic protozoan parasite Cryptosporidium parvum (CpIMPDH), which was obtained from a bacterial source through horizontal gene transfer, and its host counterpart, human IMPDH type 2 (hIMPDH2). Interestingly, the intrinsic binding energy of NAD+ differentially distributes across the dinucleotide binding sites of these two enzymes as well as in the previously characterized IMPDH from Tritrichomonas foetus (TfIMPDH). Both the dehydrogenase and hydrolase reactions display significant differences in the host and parasite enzymes, in keeping with the phylogenetic and structural divergence of their active sites. Despite large differences in Kc, the catalytic power of both the dehydrogenase and hydrolase conformations are similar in CpIMPDH and TfIMPDH. This observation suggests that the closure of the flap simply sets the stage for catalysis rather than plays a more active role in the chemical transformation. This work provides the essential mechanistic framework for drug discovery.
Co-reporter:Lizbeth Hedstrom, Lu Gan
Current Opinion in Chemical Biology 2006 Volume 10(Issue 5) pp:520-525
Publication Date(Web):October 2006
DOI:10.1016/j.cbpa.2006.08.005
Textbooks describe enzymes as relatively rigid templates for the transition state of a chemical reaction, and indeed an enzyme such as chymotrypsin, which catalyzes a relatively simple hydrolysis reaction, is reasonably well described by this model. Inosine monophosphate dehydrogenase (IMPDH) undergoes a remarkable array of conformational transitions in the course of a complicated catalytic cycle, offering a dramatic counterexample to this view. IMPDH displays several other unusual mechanistic features, including an Arg residue that may act as a general base catalyst and a dynamic monovalent cation site. Further, IMPDH appears to be involved in ’moon-lighting‘ functions that may require additional conformational states. How the balance between conformational states is maintained and how the various conformational states interconvert is only beginning to be understood.
Co-reporter:Lizbeth Hedstrom
Trends in Parasitology (September 2015) Volume 31(Issue 9) pp:401-402
Publication Date(Web):1 September 2015
DOI:10.1016/j.pt.2015.08.003
The child-killer and famously recalcitrant parasite Cryptosporidium is the latest organism to yield to the magic of CRISPR/Cas9. The ability to knockout genes and introduce markers promises a new heyday for drug discovery and vaccine development as well as the basic biology of these fascinating parasites.
Co-reporter:Dong Xu, Garrett Cobb, Catherine J. Spellicy, Sara J. Bowne, Stephen P. Daiger, Lizbeth Hedstrom
Archives of Biochemistry and Biophysics (15 April 2008) Volume 472(Issue 2) pp:100-104
Publication Date(Web):15 April 2008
DOI:10.1016/j.abb.2008.02.012
.jpg)






![(E)-3-(6-bromopyridin-2-yl)-2-cyano-N-[(1S)-1-phenylbutyl]prop-2-enamide](http://img.cochemist.com/ccimg/856300/856243-80-6.png)
![(E)-3-(6-bromopyridin-2-yl)-2-cyano-N-[(1S)-1-phenylbutyl]prop-2-enamide](http://img.cochemist.com/ccimg/856300/856243-80-6_b.png)











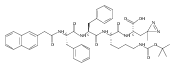
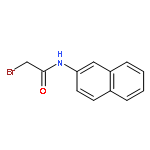
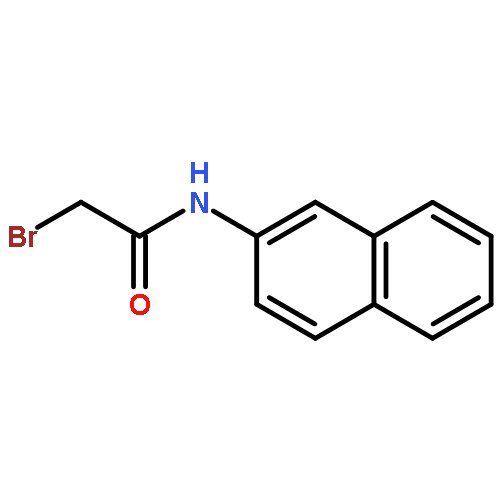
![L-Lysine, N-[2-(2-naphthalenyl)acetyl]-L-phenylalanyl-L-phenylalanyl-N6-[(1,1-dimethylethoxy)carbonyl]-](/data/chemimg/190400/1392783-93-5.png)
![L-Lysine, N-[2-(2-naphthalenyl)acetyl]-L-phenylalanyl-L-phenylalanyl-N6-[(1,1-dimethylethoxy)carbonyl]-](/data/chemimg/190400/1392783-93-5_b.png)
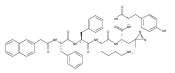

![Glycine, L-γ-glutamyl-S-[1-[2-(3,4-dihydroxyphenyl)ethyl]-2,5-dioxo-3-pyrrolidinyl]-L-cysteinyl-](/data/chemimg/190400/1388830-25-8.png)
![Glycine, L-γ-glutamyl-S-[1-[2-(3,4-dihydroxyphenyl)ethyl]-2,5-dioxo-3-pyrrolidinyl]-L-cysteinyl-](/data/chemimg/190400/1388830-25-8_b.png)































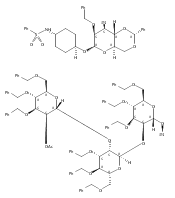


![Acetamide, 2-[4-[(2,4-diamino-5-pyrimidinyl)methyl]-2,6-dimethoxyphenoxy]-N-[2-(3,4-dihydroxyphenyl)ethyl]-](/data/chemimg/190400/1311968-22-5.png)
![Acetamide, 2-[4-[(2,4-diamino-5-pyrimidinyl)methyl]-2,6-dimethoxyphenoxy]-N-[2-(3,4-dihydroxyphenyl)ethyl]-](/data/chemimg/190400/1311968-22-5_b.png)











![Acetamide, 2-bromo-N-[4-(methylthio)phenyl]-](http://img.cochemist.com/ccimg/150300/150221-08-2.png)
![Acetamide, 2-bromo-N-[4-(methylthio)phenyl]-](http://img.cochemist.com/ccimg/150300/150221-08-2_b.png)
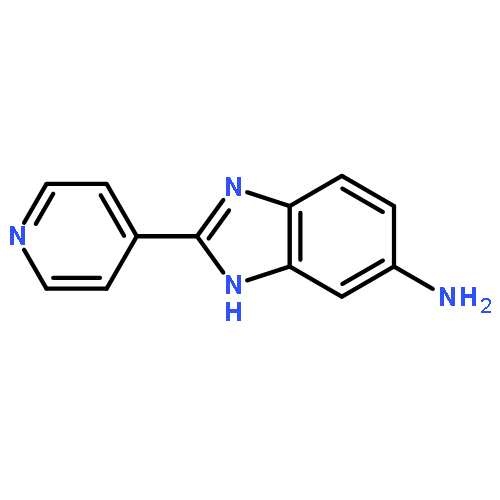
![5-Nitro-2-(pyridin-4-yl)-1H-benzo[d]imidazole](http://img.cochemist.com/ccimg/148600/148533-73-7.png)
![5-Nitro-2-(pyridin-4-yl)-1H-benzo[d]imidazole](http://img.cochemist.com/ccimg/148600/148533-73-7_b.png)








![Ethanone, 1-[3-(1-hydroxy-1-methylethyl)phenyl]-](http://img.cochemist.com/ccimg/87800/87771-41-3.png)
![Ethanone, 1-[3-(1-hydroxy-1-methylethyl)phenyl]-](http://img.cochemist.com/ccimg/87800/87771-41-3_b.png)









![(3aR,5R,6R,6aR)-6-(benzyloxy)-5-(benzyloxymethyl)-2,2-dimethyl-tetrahydrofuro[2,3-d][1,3]dioxole](http://img.cochemist.com/ccimg/55800/55735-86-9.png)
![(3aR,5R,6R,6aR)-6-(benzyloxy)-5-(benzyloxymethyl)-2,2-dimethyl-tetrahydrofuro[2,3-d][1,3]dioxole](http://img.cochemist.com/ccimg/55800/55735-86-9_b.png)




![2-Phenylbenzo[d]oxazol-5-amine](http://img.cochemist.com/ccimg/41400/41373-37-9.png)
![2-Phenylbenzo[d]oxazol-5-amine](http://img.cochemist.com/ccimg/41400/41373-37-9_b.png)
![2-AMINO-6,7,8,9-TETRAHYDRODIBENZO[B,D]FURAN 97%](http://img.cochemist.com/ccimg/38100/38084-44-5.png)
![2-AMINO-6,7,8,9-TETRAHYDRODIBENZO[B,D]FURAN 97%](http://img.cochemist.com/ccimg/38100/38084-44-5_b.png)
![Poly[imino[1-[(4-hydroxyphenyl)methyl]-2-oxo-1,2-ethanediyl]](9CI)](http://img.cochemist.com/ccimg/31800/31724-37-5.png)
![Poly[imino[1-[(4-hydroxyphenyl)methyl]-2-oxo-1,2-ethanediyl]](9CI)](http://img.cochemist.com/ccimg/31800/31724-37-5_b.png)





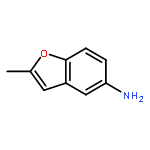
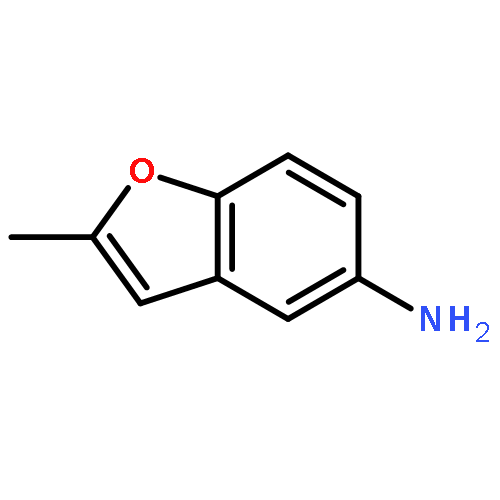

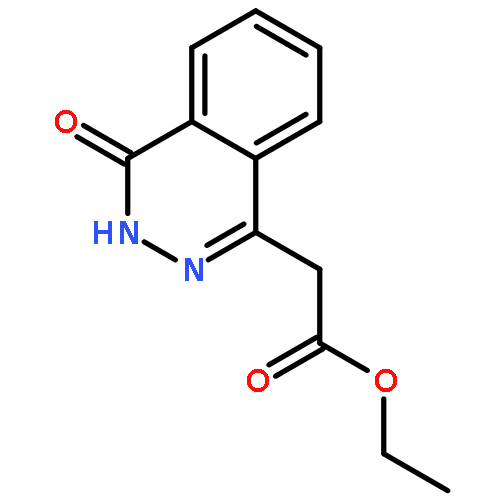
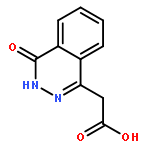











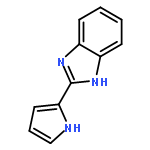
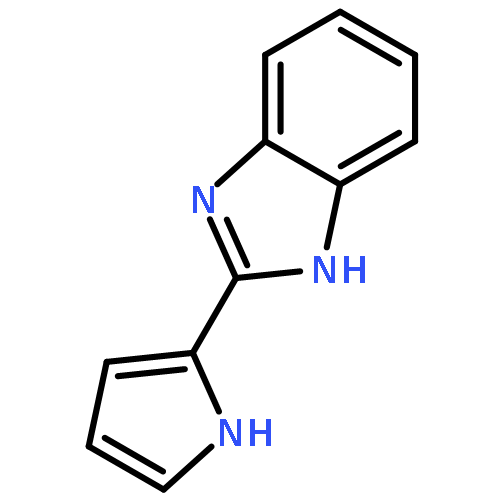
![[4-(CYCLOPROPYLACETYL)PHENYL]BORONIC ACID](http://img.cochemist.com/ccimg/3900/3878-18-0.png)
![[4-(CYCLOPROPYLACETYL)PHENYL]BORONIC ACID](http://img.cochemist.com/ccimg/3900/3878-18-0_b.png)
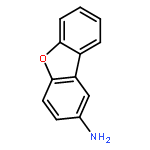
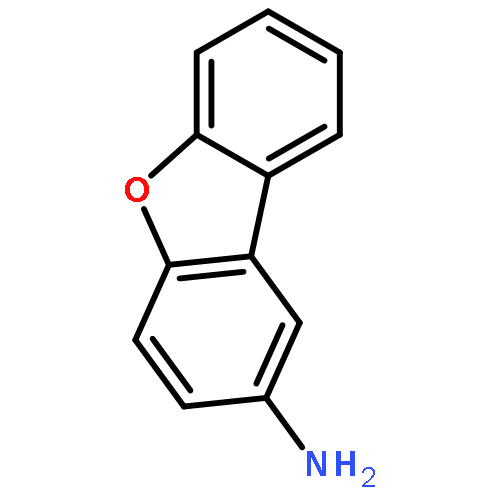



![BENZENE, [(1-METHYL-2-PROPYNYL)OXY]-](http://img.cochemist.com/ccimg/1600/1596-40-3.png)
![BENZENE, [(1-METHYL-2-PROPYNYL)OXY]-](http://img.cochemist.com/ccimg/1600/1596-40-3_b.png)







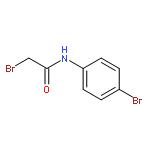
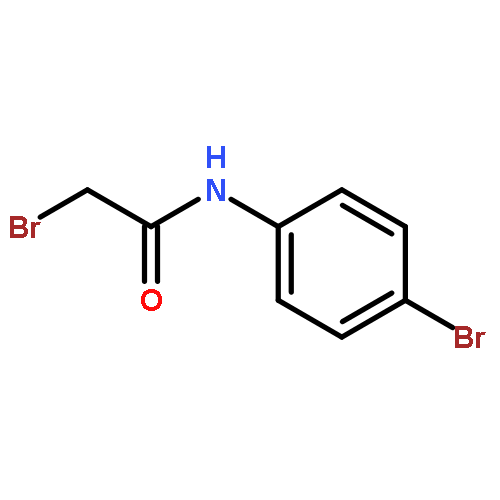
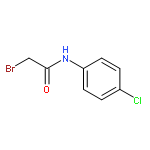
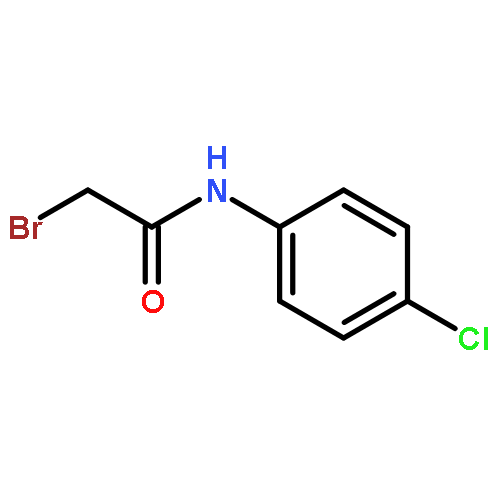




![Urea, N-[1-methyl-1-[3-(1-methylethenyl)phenyl]ethyl]-N'-phenyl-](http://img.cochemist.com/ccimg/393600/393516-98-8.png)
![Urea, N-[1-methyl-1-[3-(1-methylethenyl)phenyl]ethyl]-N'-phenyl-](http://img.cochemist.com/ccimg/393600/393516-98-8_b.png)