Co-reporter:Kyle M. Lambert, Zachary D. Stempel, Sadie M. Kiendzior, Ashley L. Bartelson, and William F. Bailey
The Journal of Organic Chemistry November 3, 2017 Volume 82(Issue 21) pp:11440-11440
Publication Date(Web):October 2, 2017
DOI:10.1021/acs.joc.7b01965
The multigram preparation and characterization of a novel TEMPO-based oxoammonium salt, 2,2,6,6-tetramethyl-4-(2,2,2-trifluoroacetamido)-1-oxopiperidinium tetrafluoroborate (5), and its corresponding nitroxide (4) are reported. The solubility profile of 5 in solvents commonly used for alcohol oxidations differs substantially from that of Bobbitt’s salt, 4-acetamido-2,2,6,6-tetramethyl-1-oxopiperidinium tetrafluoroborate (1). The rates of oxidation of a representative series of primary, secondary, and benzylic alcohols by 1 and 5 in acetonitrile solvent at room temperature have been determined, and oxoammonium salt 5 has been found to oxidize alcohols more rapidly than does 1. The rate of oxidation of meta- and para-substituted benzylic alcohols by either 1 or 5 displays a strong linear correlation to Hammett parameters (r > 0.99) with slopes (ρ) of −2.7 and −2.8, respectively, indicating that the rate-limiting step in the oxidations involves hydride abstraction from the carbinol carbon of the alcohol substrate.
Co-reporter:William F. Bailey and Justin D. Fair
The Journal of Organic Chemistry 2016 Volume 81(Issue 9) pp:3951-3955
Publication Date(Web):April 13, 2016
DOI:10.1021/acs.joc.6b00496
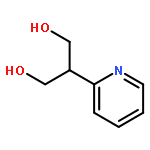
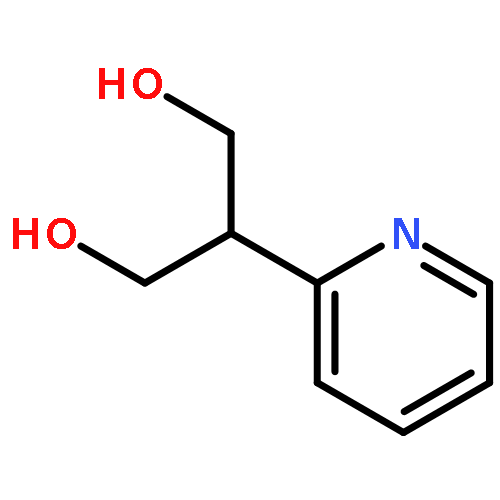
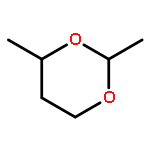
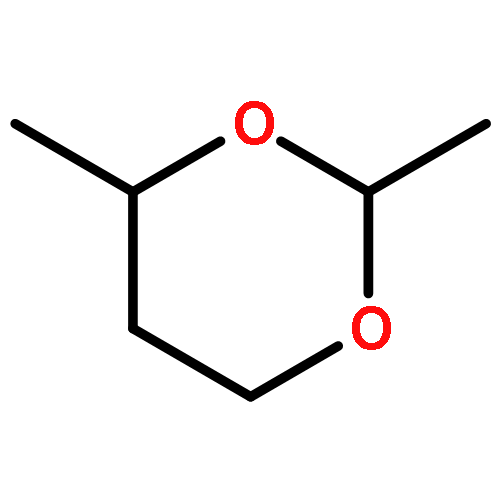
Lithium–iodine exchange-initiated fragmentation of a series of 4-substituted 2-iodomethyl-1,3-dioxanes proceeds rapidly and regioselectively to afford enol ether alcohols by preferential cleavage of the less congested C(2)–O(1) bond. The results demonstrate that a complex-induced proximity effect (CIPE) is likely responsible for the selectivity of the cleavage.
Co-reporter:Kyle M. Lambert;Dr. James M. Bobbitt;Sherif A. Eldirany;Liam E. Kissane;Rose K. Sheridan;Zachary D. Stempel;Francis H. Sternberg ;Dr. William F. Bailey
Chemistry - A European Journal 2016 Volume 22( Issue 15) pp:5156-5159
Publication Date(Web):
DOI:10.1002/chem.201600549
Abstract
Synergism among several intertwined catalytic cycles allows for selective, room temperature oxidation of primary amines to the corresponding nitriles in 85–98 % isolated yield. This metal-free, scalable, operationally simple method employs a catalytic quantity of 4-acetamido-TEMPO (ACT; TEMPO=2,2,6,6-tetramethylpiperidine N-oxide) radical and the inexpensive, environmentally benign triple salt oxone as the terminal oxidant under mild conditions. Simple filtration of the reaction mixture through silica gel affords pure nitrile products.
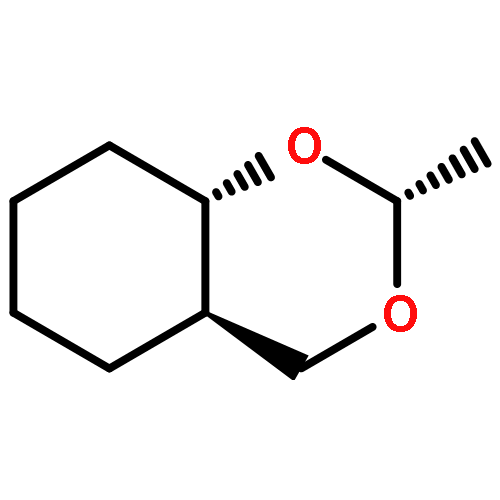
Co-reporter:William F. BaileyKyle M. Lambert, Zachary D. StempelKenneth B. Wiberg, Brandon Q. Mercado
The Journal of Organic Chemistry 2016 Volume 81(Issue 24) pp:12116-12127
Publication Date(Web):November 16, 2016
DOI:10.1021/acs.joc.6b02428
Anancomeric 5-phenyl-1,3-dioxanes provide a unique opportunity to study factors that control conformation. Whereas one might expect an axial phenyl group at C(5) of 1,3-dioxane to adopt a conformation similar to that in axial phenylcyclohexane, a series of studies including X-ray crystallography, NOE measurements, and DFT calculations demonstrate that the phenyl prefers to lie over the dioxane ring in order to position an ortho-hydrogen to participate in a stabilizing, nonclassical CH···O hydrogen bond with a ring oxygen of the dioxane. Acid-catalyzed equilibration of a series of anancomeric 2-tert-butyl-5-aryl-1,3-dioxane isomers demonstrates that remote substituents on the phenyl ring affect the conformational energy of a 5-aryl-1,3-dioxane: electron-withdrawing substituents decrease the conformational energy of the aryl group, while electron-donating substituents increase the conformational energy of the group. This effect is correlated in a very linear way to Hammett substituent parameters. In short, the strength of the CH···O hydrogen bond may be tuned in a predictable way in response to the electron-withdrawing or electron-donating ability of substituents positioned remotely on the aryl ring. This effect may be profound: a 3,5-bis-CF3 phenyl group at C(5) in 1,3-dioxane displays a pronounced preference for the axial orientation. The results are relevant to broader conformational issues involving heterocyclic systems bearing aryl substituents.
Co-reporter:Christopher B. Kelly;Kyle M. Lambert;Michael A. Mercadante;John M. Ovian; William F. Bailey; Nicholas E. Leadbeater
Angewandte Chemie International Edition 2015 Volume 54( Issue 14) pp:4241-4245
Publication Date(Web):
DOI:10.1002/anie.201412256
Abstract
A scalable, high yielding, rapid route to access an array of nitriles from aldehydes mediated by an oxoammonium salt (4-acetylamino-2,2,6,6-tetramethylpiperidine-1-oxoammonium tetrafluoroborate) and hexamethyldisilazane (HMDS) as an ammonia surrogate has been developed. The reaction likely involves two distinct chemical transformations: reversible silyl-imine formation between HMDS and an aldehyde, followed by oxidation mediated by the oxoammonium salt and desilylation to furnish a nitrile. The spent oxidant can be easily recovered and used to regenerate the oxoammonium salt oxidant.
Co-reporter:Christopher B. Kelly;Kyle M. Lambert;Michael A. Mercadante;John M. Ovian; William F. Bailey; Nicholas E. Leadbeater
Angewandte Chemie 2015 Volume 127( Issue 14) pp:4315-4319
Publication Date(Web):
DOI:10.1002/ange.201412256
Abstract
A scalable, high yielding, rapid route to access an array of nitriles from aldehydes mediated by an oxoammonium salt (4-acetylamino-2,2,6,6-tetramethylpiperidine-1-oxoammonium tetrafluoroborate) and hexamethyldisilazane (HMDS) as an ammonia surrogate has been developed. The reaction likely involves two distinct chemical transformations: reversible silyl-imine formation between HMDS and an aldehyde, followed by oxidation mediated by the oxoammonium salt and desilylation to furnish a nitrile. The spent oxidant can be easily recovered and used to regenerate the oxoammonium salt oxidant.
Co-reporter:William F. Bailey, Kyle M. Lambert, Kenneth B. Wiberg, and Brandon Q. Mercado
The Journal of Organic Chemistry 2015 Volume 80(Issue 8) pp:4108-4115
Publication Date(Web):March 24, 2015
DOI:10.1021/acs.joc.5b00422
The conformational preference of a variety of 2,2-diaryl-1,3-dioxanes bearing remote substituents on the phenyl rings has been studied via equilibration of configurationally isomeric 2,2-diaryl-cis-4,6-dimethyl-1,3-dioxane epimers, X-ray crystallography, 1H NOESY analysis, and B3LYP/6-311+G* calculations. When the aryl ring bears a remote electron-withdrawing substituent, the isomer having both the higher dipole moment and the electron-withdrawing group in the equatorial phenyl ring and/or an electron-donating group in the axial ring has the lower energy. These results differ from the conclusions reported in a previous study of similar systems. The conformational energy differences of para-substituted 2,2-diaryl-1,3-dioxanes are linearly related to the Hammett σ values with a slope (ρ) of 0.6. In addition, there is a trend toward longer bond lengths between the C(2) ketal center and the aryl ring as the electron-withdrawing nature of the para-substituent is increased. Electrostatic interactions, rather than a hyperconjugative anomeric effect, appear to be responsible for the conformational behavior of such molecules.
Co-reporter:Kenneth B. Wiberg, Kyle M. Lambert, and William F. Bailey
The Journal of Organic Chemistry 2015 Volume 80(Issue 16) pp:7884-7889
Publication Date(Web):July 16, 2015
DOI:10.1021/acs.joc.5b01340
The rotameric conformations of the phenyl ring in both the axial and the equatorial conformers of phenyl substituted 1,3-dioxanes and tetrahydropyrans are compared with those of the corresponding phenylcyclohexanes at the MP2/6-311+G* level. The compounds with an axial phenyl commonly adopt a conformation in which the plane of the aromatic ring is perpendicular to the benzylic C–H bond. However, axial 5-phenyl-1,3-dioxane adopts a “parallel” conformation that allows an ortho hydrogen to be proximate to the two ring oxygens, leading to attractive CH···O interactions. Stabilizing Coulombic interactions of this sort are found with many of the oxygen-containing six-membered rings that were investigated.
Co-reporter:Kyle M. Lambert, James M. Bobbitt, Sherif A. Eldirany, Kenneth B. Wiberg, and William F. Bailey
Organic Letters 2014 Volume 16(Issue 24) pp:6484-6487
Publication Date(Web):December 11, 2014
DOI:10.1021/ol503345h
The oxidation of primary amines using a stoichiometric quantity of 4-acetamido-2,2,6,6-tetramethylpiperidine-1-oxoammonium tetrafluoroborate (1) in CH2Cl2–pyridine solvent at room temperature or at gentle reflux affords nitriles in good yield under mild conditions. The mechanism of the oxidation, which has been investigated computationally, involves a hydride transfer from the amine to the oxygen atom of 1 as the rate-limiting step.
Co-reporter:William F. Bailey, Kenneth B. Wiberg, Ashley L. Bartelson
Tetrahedron Letters 2014 Volume 55(Issue 34) pp:4807-4809
Publication Date(Web):20 August 2014
DOI:10.1016/j.tetlet.2014.07.011
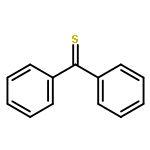
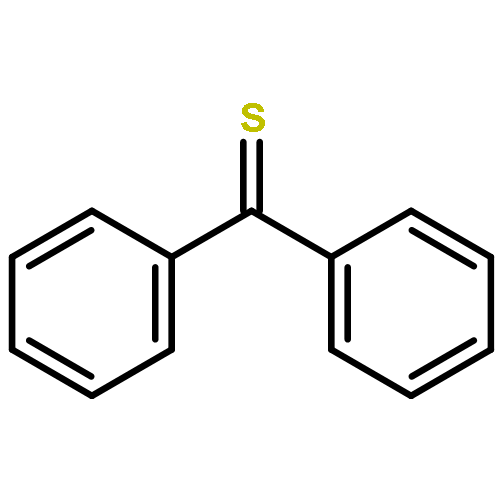
The reactions of adamantanethione (1) with several Grignard reagents (MeMgCl, n-BuMgCl, and t-BuMgCl) have been investigated. In contrast to analogous reactions with ketones, there is no evidence of addition to the CS carbon of 1. Rather, reaction of 1 with n-BuMgCl or t-BuMgCl affords 2-adamantanethiol as the major product along with minor amounts of sulfides and an episulfide. Reactions of 1 with MeMgCl are very slow and afford a variety of products as a result of secondary reactions.
Co-reporter:Matthew R. Luderer, Michael J. Mealy, and William F. Bailey
The Journal of Organic Chemistry 2014 Volume 79(Issue 21) pp:10722-10726
Publication Date(Web):October 10, 2014
DOI:10.1021/jo502139m
Concise syntheses of the sesquiterpenes (R)-(+)-cuparene and (R)-(+)-herbertene by asymmetric cyclization of achiral olefinic alkyllithium precursors in the presence of (−)-sparteine are reported. The quaternary stereogenic center in each product is set at the final step of the synthesis by enantioselective (er = 61:39) 5-exo ring closure.
Co-reporter:William F. Bailey and Johanna M. Bakonyi
The Journal of Organic Chemistry 2013 Volume 78(Issue 7) pp:3493-3495
Publication Date(Web):March 7, 2013
DOI:10.1021/jo4002118
A concise preparation of the pheromone secreted by the female longtailed mealybug [viz., 2-(1,5,5-trimethylcyclopent-2-en-1-yl)ethyl acetate] (1) is described. The key step in the synthesis of 1 involves 5-exo-trig ring closure of the vinyllithium derived from (Z)-1-iodo-4,4,5-trimethyl-1,5-hexadiene by lithium–iodine exchange.
Co-reporter:William F. Bailey ; Ashley L. Bartelson ;Kenneth B. Wiberg
Journal of the American Chemical Society 2012 Volume 134(Issue 6) pp:3199-3207
Publication Date(Web):January 13, 2012
DOI:10.1021/ja210847n
The reaction of ketones with organolithium reagents generally proceeds by addition of the organometallic to the electrophilic carbon of the C═O group to give the lithium salt of a tertiary alcohol. The seemingly analogous reaction of thioketones with organolithiums is a fundamentally different process: such reactions typically afford a variety of products, and addition of the organolithium to carbon of the C═S group to give a thiol is a relatively unimportant component. Reactions of the stable thioketone, adamantantanethione (1), with several alkyllithiums (MeLi, n-BuLi and t-BuLi) in a variety of solvents have been studied in the first comprehensive investigation of the reactions of organolithiums with a representative alkyl-substituted thione. Reactions of 1 with n-BuLi or t-BuLi afforded 2-adamantanethiol (2) as the major product. In an effort to explain the marked difference in behavior of ketones and thioketones in reactions with organolithiums, transition states for both the addition and reduction reactions have been located at the B3LYP/6-311+G* level using acetone and thioacetone as model substrates. The transition states for the addition of dimeric MeLi to the C═O and C═S carbons of acetone and thioacetone were significantly different as a result of the small bond angles preferred by divalent sulfur, and this accounts for the much slower addition to a C═S carbon vis-à-vis a C═O group. Transition states for reduction of acetone and thioacetone by EtLi were similar, but the greater exothermicity of the reduction of the thioketone results in an earlier transition state and lower activation energy for this process than that for the reduction of a ketone. The possible role of radical-mediated processes in this chemistry is also discussed.
Co-reporter:Joseph C. Qiu, Priya P. Pradhan, Nyle B. Blanck, James M. Bobbitt, and William F. Bailey
Organic Letters 2012 Volume 14(Issue 1) pp:350-353
Publication Date(Web):December 8, 2011
DOI:10.1021/ol203096f
The oxidation of alcohols to aldehydes using stoichiometric 4-acetamido-2,2,6,6-tetramethylpiperidine-1-oxoammonium tetrafluoroborate (1) in CH2Cl2 at room temperature is a highly selective process favoring reaction at the carbinol center best able to accommodate a positive charge. The oxidation of aldehydes to carboxylic acids by 1 in wet acetonitrile is also selective; the rate of the process correlates with the concentration of aldehyde hydrate. A convenient and high yield method for oxidation of alcohols directly to carboxylic acids has been developed.
Co-reporter:William F. Bailey, Mark R. Luderer, Daniel P. Uccello and Ashley L. Bartelson
The Journal of Organic Chemistry 2010 Volume 75(Issue 8) pp:2661-2666
Publication Date(Web):March 22, 2010
DOI:10.1021/jo100303q
The outcome of reactions of (E)-5-bromo-5-decene (1), a representative vinyl bromide, with t-BuLi or n-BuLi at 0 °C and room temperature, respectively, in a variety of solvent systems has been investigated. Vinyl bromide 1 does not react with t-BuLi in pure heptane; however, the presence of even small quantities of an ether in a predominantly heptane medium resulted in virtually complete consumption of 1 at 0 °C, resulting in nearly the same distribution of products, including 60−80% of (Z)-5-decenyllithium, regardless of the solvent composition. Vinyl bromide 1 reacts slowly with n-BuLi at room temperature in a variety of ether and heptane-ether mixtures to afford a mixture of products including significant quantities of recovered starting material. The results of these experiments demonstrate that lithium−bromine exchange between a vinyl bromide and either t-BuLi or n-BuLi at temperatures significantly above −78 °C is not an efficient method for the generation of a vinyllithium.
Co-reporter:Terry ‘Li’ Rathman and William F. Bailey
Organic Process Research & Development 2009 Volume 13(Issue 2) pp:144-151
Publication Date(Web):December 10, 2008
DOI:10.1021/op800246z
Over the last several decades, research directed at optimization of reactions involving organolithium reagents has led to the recognition that a variety of experimental parameters may affect the outcome and viability of such reactions. Investigation of the factors that influence organolithium-mediated reactions on a large scale is a requirement for development of a feasible and practical process. This contribution critically reviews selected examples, taken from the literature, in which adjustment of the reaction medium, order of addition, temperature, the presence of additives, and judicious choice of base, substrate and/or electrophile resulted in optimization of processes involving organolithium reactions.
Co-reporter:Kenneth B. Wiberg, Yi-gui Wang, Scott J. Miller and Angela L. A. Puchlopek, William F. Bailey and Justin D. Fair
The Journal of Organic Chemistry 2009 Volume 74(Issue 10) pp:3659-3664
Publication Date(Web):April 16, 2009
DOI:10.1021/jo9004316
The reaction of benzoyl chloride with methanol catalyzed by pyridine is 9 times more rapid than is the same reaction with thiobenzoyl chloride. The difference in reactivity, as well as the dealkylation reactions that occur when the reaction of thiobenzoyl chloride is catalyzed by bases such as Et3N, can be understood in terms of the charge distributions in the intermediate acylammonium ions. The reaction of PhNCO with ethanol occurs at a much higher rate (4.8 × 104) than that of PhNCS, corresponding to a difference in activation free energies for the additions of 6 kcal/mol. Transition states for each of these reactions were located, and each involves two alcohol molecules in a hydrogen bonded six-membered ring arrangement. Information concerning differences in reactivity was derived from analysis of Hirshfeld atomic charge distributions and calculated hydrogenolysis reaction energies.
Co-reporter:Priya P. Pradhan, James M. Bobbitt and William F. Bailey
The Journal of Organic Chemistry 2009 Volume 74(Issue 24) pp:9524-9527
Publication Date(Web):October 30, 2009
DOI:10.1021/jo902144b
Benzylic ethers and related ArCH2OR substrates are oxidatively cleaved by 4-acetamido-2,2,6,6-tetramethylpiperidine-1-oxoammonium tetrafluoroborate (1) in wet CH3CN at room temperature to give the corresponding aromatic aldehyde and alcohol in high yield. Primary or secondary alcohol products are further oxidized by 1 to give carboxylic acids and ketones, respectively. The oxidation likely involves a formal hydride abstraction from the benzylic carbon as evidenced by slow reaction of substrates bearing electron-withdrawing substituents.
Co-reporter:Matthew R. Luderer, William F. Bailey, Mark R. Luderer, Justin D. Fair, Robert J. Dancer, Michael Bech Sommer
Tetrahedron: Asymmetry 2009 Volume 20(Issue 9) pp:981-998
Publication Date(Web):21 May 2009
DOI:10.1016/j.tetasy.2009.03.015
The enantioselective preparation of chiral secondary and tertiary alcohols via addition of an achiral organomagnesium reagent or an organolithium to an achiral aldehyde or ketone in a chiral medium is reviewed. The review is written in chronological order and contains 113 references to literature through late 2008.
.jpg)

![Cyclohexanol, 2-[(ethenyloxy)methyl]-, trans-](http://img.cochemist.com/ccimg/142200/142178-88-9.png)
![Cyclohexanol, 2-[(ethenyloxy)methyl]-, trans-](http://img.cochemist.com/ccimg/142200/142178-88-9_b.png)
![1-Propanamine, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/115400/115306-75-7.png)
![1-Propanamine, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/115400/115306-75-7_b.png)
![Propanenitrile, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/90000/89923-33-1.png)
![Propanenitrile, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/90000/89923-33-1_b.png)




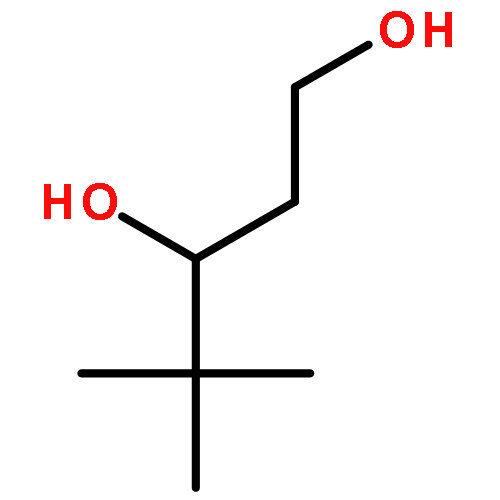
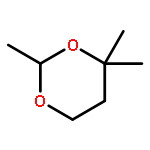
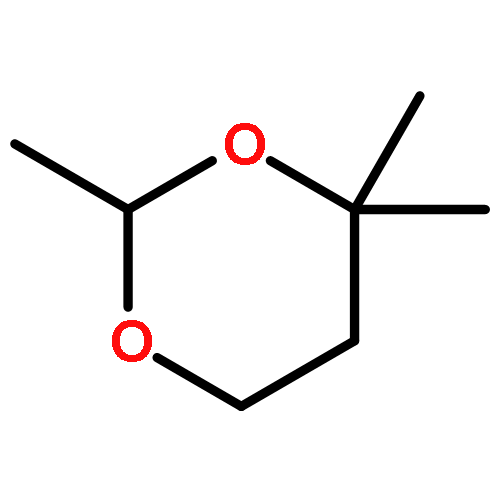
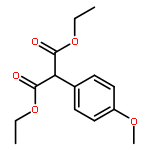

![1-Butanol, 3-[(acetyloxy)methoxy]-3-methyl-, acetate](http://img.cochemist.com/ccimg/16700/16681-24-6.png)
![1-Butanol, 3-[(acetyloxy)methoxy]-3-methyl-, acetate](http://img.cochemist.com/ccimg/16700/16681-24-6_b.png)











![DISPIRO[TRICYCLO[3.3.1.13,7]DECANE-2,2'-THIIRANE-3',2''-TRICYCLO[3.3.1.13,7]DECANE]](http://img.cochemist.com/ccimg/62300/62281-75-8.png)
![DISPIRO[TRICYCLO[3.3.1.13,7]DECANE-2,2'-THIIRANE-3',2''-TRICYCLO[3.3.1.13,7]DECANE]](http://img.cochemist.com/ccimg/62300/62281-75-8_b.png)



















![Benzo[d][1,3]dioxole-5-carbonitrile](http://img.cochemist.com/ccimg/4500/4421-09-4.png)
![Benzo[d][1,3]dioxole-5-carbonitrile](http://img.cochemist.com/ccimg/4500/4421-09-4_b.png)












![3-[4-(HYDROXYMETHYL)PHENOXY]PROPAN-1-OL](http://img.cochemist.com/ccimg/341100/341035-93-6.png)
![3-[4-(HYDROXYMETHYL)PHENOXY]PROPAN-1-OL](http://img.cochemist.com/ccimg/341100/341035-93-6_b.png)




![METHYL (1S,4AS,5R,7S,7AS)-7-ACETOXY-1-(WEI -D-GLUCOPYRANOSYLOXY)-5-H<WBR />YDROXY-7-METHYL-1,4A,5,6,7,7A-HEXAHYDROCYCLOPENTA[C]PYRAN-4-CARBO<WBR />XYLATE](http://img.cochemist.com/ccimg/38700/38628-53-4.png)
![METHYL (1S,4AS,5R,7S,7AS)-7-ACETOXY-1-(WEI -D-GLUCOPYRANOSYLOXY)-5-H<WBR />YDROXY-7-METHYL-1,4A,5,6,7,7A-HEXAHYDROCYCLOPENTA[C]PYRAN-4-CARBO<WBR />XYLATE](http://img.cochemist.com/ccimg/38700/38628-53-4_b.png)





![Tricyclo[3.3.1.13,7]decanethione](http://img.cochemist.com/ccimg/23700/23695-65-0.png)
![Tricyclo[3.3.1.13,7]decanethione](http://img.cochemist.com/ccimg/23700/23695-65-0_b.png)