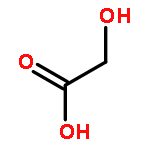
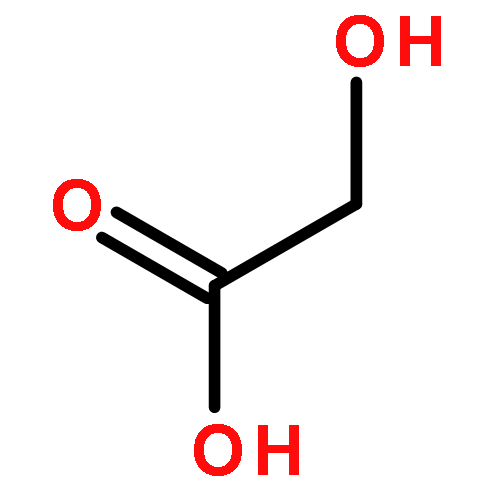
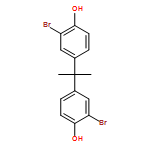
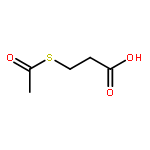
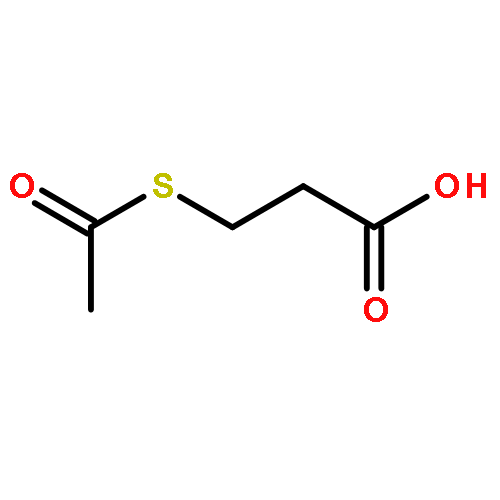
Co-reporter:Laura Bertoletti, Julie Schappler, Raffaella Colombo, Serge Rudaz, Rob Haselberg, Elena Domínguez-Vega, Sara Raimondi, Govert W. Somsen, Ersilia De Lorenzi
Analytica Chimica Acta 2016 Volume 945() pp:102-109
Publication Date(Web):16 November 2016
DOI:10.1016/j.aca.2016.10.010
•CE-ESI-MS is proposed for protein conformational analysis.•The amyloidogenic protein beta2-microglobulin is used as a model.•Protein conformers are studied under native and misfolding conditions.•New sheathless interfacing is best suited to preserve protein structure integrity.•CE-ESI-TOF MS reliably assigns protein forms that differ by 1 Da.In this work we explored the feasibility of different CE-ESI-MS set-ups for the analysis of conformational states of an intact protein. By using the same background electrolyte at quasi physiological conditions (50 mM ammonium bicarbonate, pH 7.4) a sequential optimization was carried out, initially by evaluating a sheath-liquid interface with both a single quadrupole (SQ) and a time-of-flight (TOF) mass spectrometer; then a sheathless interface coupled with high-resolution QTOF MS was considered. Beta2-microglobulin has been taken as a model, as it is an amyloidogenic protein and its conformational changes are strictly connected to the onset of a disease. The separation of two conformers at dynamic equilibrium is achieved all the way down to the MS detection. Notably, the equilibrium ratio of the protein conformers is maintained in the electrospray source after CE separation. Strengths and weaknesses of each optimized set-up are emphasized and their feasibility in unfolding studies is evaluated. In particular, ESI-TOF MS can assign protein forms that differ by 1 Da only and sheathless interfacing is best suited to preserve protein structure integrity. This demonstrates the CE-ESI-MS performance in terms of separation, detection and characterization of conformational species that co-populate a protein solution.
Co-reporter:Bregje J. de Kort, Gerhardus J. de Jong and Govert W. Somsen
Analyst 2013 vol. 138(Issue 16) pp:4550-4557
Publication Date(Web):09 May 2013
DOI:10.1039/C3AN00357D
Capillary electrophoresis (CE) with wavelength-resolved fluorescence detection (wrFlu) was evaluated for the study of protein unfolding using non-reduced and reduced β-lactoglobulin B (β-LGB) as model compounds. Protein unfolding was achieved by incubation in sodium phosphate (pH 3.0) containing increasing concentrations of urea (0–7.1 M). CE-wrFlu was performed using the incubation media as background electrolytes (BGEs). At low urea concentrations (0–3.1 M), CE-wrFlu analysis of non-reduced β-LGB showed a single peak with a maximum emission wavelength (λmax) of 333 nm, indicating the protein was in its folded state. When β-LGB was exposed to 3.6 and 4.1 M urea, CE-wrFlu revealed an additional peak with a λmax of 353 nm and a reduced migration time, suggesting the formation of fully unfolded species. Upon further raising the urea concentration up to 6.5 M, the peak intensity of the unfolded protein increased. At the same time, the later-migrating peak became wider and lower, showing a decrease of migration time and a shift of λmax (333–353 nm), indicating gradual unfolding. Construction of a λmax-based transition curve for the later-migrating β-LGB species provided a denaturant-concentration midpoint of unfolding (cm) of 5.39 M, which was similar to the cm determined by plotting the corrected effective electrophoretic mobility (μeff,c) vs. urea concentration. Stand-alone fluorescence spectroscopy of the same β-LGB solutions revealed a lower cm (4.97 M), most probably because the respective β-LGB species were not separated, yielding ensemble average data. For reduced β-LGB, at all tested urea concentrations one protein peak was observed, whereas λmax and μeff,c indicated protein unfolding at much lower urea concentrations (cm of 1.2 M). We conclude that CE-wrFlu can distinguish protein conformational states and thus may provide useful additional information on unfolding pathways.













![4-AMINO-N-1-AZABICYCLO[2.2.2]OCT-3-YL-5-CHLORO-2-METHOXYBENZAMIDE HYDROCHLORIDE](http://img.cochemist.com/ccimg/90200/90182-92-6.png)
![4-AMINO-N-1-AZABICYCLO[2.2.2]OCT-3-YL-5-CHLORO-2-METHOXYBENZAMIDE HYDROCHLORIDE](http://img.cochemist.com/ccimg/90200/90182-92-6_b.png)


![Benzamide,N-(3R)-1-azabicyclo[2.2.2]oct-3-yl-4-chloro-, hydrochloride (1:1)](http://img.cochemist.com/ccimg/123500/123464-89-1.png)
![Benzamide,N-(3R)-1-azabicyclo[2.2.2]oct-3-yl-4-chloro-, hydrochloride (1:1)](http://img.cochemist.com/ccimg/123500/123464-89-1_b.png)


![Phenol,2,6-dibromo-4-[1-(3-bromo-4-hydroxyphenyl)-1-methylethyl]-](http://img.cochemist.com/ccimg/6400/6386-73-8.png)
![Phenol,2,6-dibromo-4-[1-(3-bromo-4-hydroxyphenyl)-1-methylethyl]-](http://img.cochemist.com/ccimg/6400/6386-73-8_b.png)
![1H-1,4,7-Triazonine-1,4,7-triaceticacid, hexahydro-2-[(4-isothiocyanatophenyl)methyl]-](http://img.cochemist.com/ccimg/147600/147597-66-8.png)
![1H-1,4,7-Triazonine-1,4,7-triaceticacid, hexahydro-2-[(4-isothiocyanatophenyl)methyl]-](http://img.cochemist.com/ccimg/147600/147597-66-8_b.png)
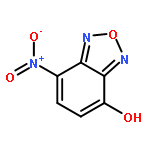
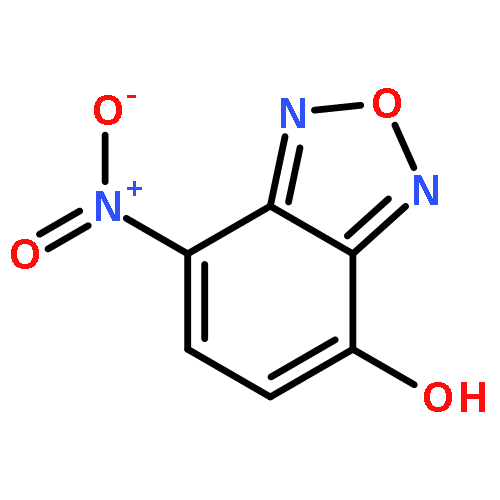
![4-Nitrobenzo[c][1,2,5]oxadiazole](http://img.cochemist.com/ccimg/16400/16322-19-3.png)
![4-Nitrobenzo[c][1,2,5]oxadiazole](http://img.cochemist.com/ccimg/16400/16322-19-3_b.png)















![2-[4,7-BIS(CARBOXYMETHYL)-1,4,7-TRIAZONAN-1-YL]ACETIC ACID](http://img.cochemist.com/ccimg/56500/56491-86-2.png)
![2-[4,7-BIS(CARBOXYMETHYL)-1,4,7-TRIAZONAN-1-YL]ACETIC ACID](http://img.cochemist.com/ccimg/56500/56491-86-2_b.png)






![Carbamimidothioic acid,N-[2-[4-(iodo-125I)phenyl]ethyl]-, 3-(1H-imidazol-5-yl)propyl ester](http://img.cochemist.com/ccimg/143500/143407-29-8.png)
![Carbamimidothioic acid,N-[2-[4-(iodo-125I)phenyl]ethyl]-, 3-(1H-imidazol-5-yl)propyl ester](http://img.cochemist.com/ccimg/143500/143407-29-8_b.png)




![1H-Indole-3-acetamide,N-1-azabicyclo[2.2.2]oct-3-yl-1-methyl-a-oxo-](http://img.cochemist.com/ccimg/143200/143137-35-3.png)
![1H-Indole-3-acetamide,N-1-azabicyclo[2.2.2]oct-3-yl-1-methyl-a-oxo-](http://img.cochemist.com/ccimg/143200/143137-35-3_b.png)



![4-Nitro-7-(piperazin-1-yl)benzo[c][1,2,5]oxadiazole](/data/chemimg/461700/139332-66-4.png)
![4-Nitro-7-(piperazin-1-yl)benzo[c][1,2,5]oxadiazole](/data/chemimg/461700/139332-66-4_b.png)