Co-reporter:Amanda B. Hummon;Xiao-Shan Yue
Journal of Proteome Research September 6, 2013 Volume 12(Issue 9) pp:4176-4186
Publication Date(Web):2017-2-22
DOI:10.1021/pr4005234
Typical mass spectrometric phosphoproteome studies are complicated by the need for large amounts of starting material and extensive sample preparation to ensure sufficient phosphopeptide identifications. In this paper, we present a novel strategy to perform optimized multistep IMAC enrichment from whole cell lysates followed by high-pH reverse phase fractionation (multi-IMAC-HLB; HLB means hydrophilic–lipophilic-balanced reversed-phase cartridge). The peptide-to-IMAC ratio was optimized to maximize IMAC performance, while multistep IMAC enrichment enabled improved phosphopeptide acquisition. The addition of the HLB step further fractionates the IMAC enriched phosphopeptides while desalting the samples, which dramatically reduces the sample manipulation time and sample loss compared to other popular strategies. We compared the phosphopeptide identification results of the multi-IMAC-HLB method with 3 mg of starting material to the well-established SCX-IMAC method with 15 mg of starting material. We identified 8969 unique phosphopeptides with the multi-IMAC-HLB method, compared to 5519 unique phosphopeptides identified with the SCX-IMAC method, an increase of 62.5%. The increase in the numbers of identified phosphopeptides is due to the increase in the ratio of identified phosphopeptides out of all detected peptides, 70.5% with multi-IMAC-HLB method compared to 32.3% with the SCX-IMAC method. Multi-IMAC-HLB is a robust and efficient method for in-depth phosphoproteomic research.Keywords: high pI phosphopeptides; high-pH reverse phase fractionation; multistep IMAC enrichment; phosphoproteomics; SCX fractionation;
Co-reporter:Monica M. Schroll, Gabriel J. LaBonia, Katelyn R. Ludwig, and Amanda B. Hummon
Journal of Proteome Research August 4, 2017 Volume 16(Issue 8) pp:3009-3009
Publication Date(Web):June 26, 2017
DOI:10.1021/acs.jproteome.7b00293
Drug resistance is a prevalent phenomenon that decreases the efficacy of cancer treatments and contributes to cancer progression and metastasis. Weakening drug-resistant cancer cells prior to chemotherapy is a potential strategy to combat chemoresistance. One approach to damage resistant cancer cells is modulation of nutritional intake. The combination of nutrient restriction with targeted compound treatment results in pronounced molecular changes. This study provides valuable information about augmenting existing chemotherapeutic regimes with simultaneous glucose restriction and autophagy inhibition in colorectal cancer cells. In this study, we explore the chemical pathways that drive the cellular response to nutrient restriction, autophagy inhibition, and the chemotherapy irinotecan using global quantitative proteomics and imaging mass spectrometry. We determined that significant pathways were altered including autophagy and metabolism via glycolysis, gluconeogenesis, and sucrose degradation. We also found that period circadian clock 2 (PER2), a tumor suppressor protein, was significantly up-regulated only when glucose was restricted with autophagy inhibition and chemotherapy. The upstream regulators of these differentially regulated pathways were determined to have implications in cancer, showing an increase in tumor suppressor proteins and a decrease in nuclear protein 1 (NUPR1) an important protein in chemoresistance. We also evaluated the phenotypic response of these cells and discovered autophagy inhibition and chemotherapy treatment increased apoptosis and decreased cell clonogenicity and viability. When glucose restriction was combined with autophagy inhibition and chemotherapy, all of the phenotypic results were intensified. In sum, our results indicate that glucose metabolism is of great importance in the ability of cancer cells to survive chemotherapy. By weakening cancer cells with glucose restriction and autophagy inhibition prior to chemotherapy, cancer cells become more sensitive to therapy.Keywords: autophagy; chloroquine; colorectal cancer; iTRAQ; MALDI imaging mass spectrometry; nutrient restriction; proteomics; three-dimensional cell culture;
Co-reporter:Jessica K. Lukowski, Eric M. Weaver, and Amanda B. Hummon
Analytical Chemistry August 15, 2017 Volume 89(Issue 16) pp:8453-8453
Publication Date(Web):July 21, 2017
DOI:10.1021/acs.analchem.7b02006
Cancer chemotherapeutics often fail to reach all diseased cells. To help solve this problem, researchers are investigating novel drug delivery systems. Liposomes are an attractive option due to their low toxicity, high biocompatibility, and potential to carry a large amount of a drug to the tumor site, all while avoiding being eliminated from the body. This study evaluates the penetration of doxorubicin-encased liposomes into three-dimensional cell cultures, or spheroids. Liposomes composed of lipids containing head groups of phosphatidylcholine (PC), phosphatidylethanolamine (PE), and cholesterol were created by extrusion. Doxorubicin is encapsulated within the hydrophilic core of the liposome. The drug is actively released in the spheroid as the lipids bind to cellular lipid bilayers. Spheroids were dosed with liposomal doxorubicin, free doxorubicin, or media control to assess drug distribution over the course of 72 h. Drug penetration was visualized by Matrix-Assisted Laser Desorption/Ionization–Imaging Mass Spectrometry (MALDI–IMS) with confirmation by steady state fluorescence microscopy, creating a comprehensive picture of drug distribution. This technique is able to identify both free and liposomal doxorubicin throughout the spheroid after just 12 hours of treatment. Additionally, MALDI–IMS is able to detect three metabolites of doxorubicin, indicating that cells actively metabolize the drug during treatment. Steady state fluorescence microscopy cannot distinguish the drug from its metabolites as they have the same emission spectra. This report summarizes the first study to use MALDI–IMS to analyze drug penetration of a liposomal drug carrier as well as its metabolites.
Co-reporter:Katelyn R. Ludwig
Molecular BioSystems (2005-Present) 2017 vol. 13(Issue 4) pp:648-664
Publication Date(Web):2017/03/28
DOI:10.1039/C6MB00656F
Sepsis is a serious medical condition that occurs in 30% of patients in intensive care units (ICUs). Early detection of sepsis is key to prevent its progression to severe sepsis and septic shock, which can cause organ failure and death. Diagnostic criteria for sepsis are nonspecific and hinder a timely diagnosis in patients. Therefore, there is currently a large effort to detect biomarkers that can aid physicians in the diagnosis and prognosis of sepsis. Mass spectrometry is often the method of choice to detect metabolomic and proteomic changes that occur during sepsis progression. These “omics” strategies allow for untargeted profiling of thousands of metabolites and proteins from human biological samples obtained from septic patients. Differential expression of or modifications to these metabolites and proteins can provide a more reliable source of diagnostic biomarkers for sepsis. Here, we focus on the current knowledge of biomarkers of sepsis and discuss the various mass spectrometric technologies used in their detection. We consider studies of the metabolome and proteome and summarize information regarding potential biomarkers in both general and neonatal sepsis.
Co-reporter:Alissa J. Schunter;Xiaoshan Yue
Analytical and Bioanalytical Chemistry 2017 Volume 409( Issue 7) pp:1749-1763
Publication Date(Web):2017 March
DOI:10.1007/s00216-016-0125-5
The contributions of phosphorylation-mediated signaling networks to colon cancer metastasis are poorly defined. To interrogate constitutive signaling alterations in cancer progression, the global phosphoproteomes of patient-matched SW480 (primary colon tumor origin) and SW620 (lymph node metastasis) cell lines were compared with TiO2 and immobilized metal affinity chromatography phosphopeptide enrichment followed by liquid chromatography–tandem mass spectrometry. Network analysis of the significantly altered phosphosites revealed differential regulation in cellular adhesion, mitosis, and messenger RNA translational machinery. Messenger RNA biogenesis and splicing, transport through the nuclear pores, initiation of translation, and stability and degradation were also affected. Although alterations in these processes have been associated with oncogenic transformation, control of messenger RNA stability has typically not been associated with cancer progression. Notably, the single phosphosite with the greatest relative change in SW620 cells was Ser2 on eukaryotic translation initiation factor 2 subunit 2, suggesting that SW620 cells translate faster or with greater efficiency than SW480 cells. These broad changes in the regulation of translation also occur without overexpression of eukaryotic translation initiation factor 4E. The findings suggest that metastatic cells exhibit constitutive changes to the phosphoproteome, and that messenger RNA stability and translational efficiency may be important targets of deregulation during cancer progression.
Co-reporter:Peter E. Feist, Simone Sidoli, Xin Liu, Monica M. Schroll, Sharif Rahmy, Rina Fujiwara, Benjamin A. Garcia, and Amanda B. Hummon
Analytical Chemistry 2017 Volume 89(Issue 5) pp:
Publication Date(Web):February 9, 2017
DOI:10.1021/acs.analchem.6b03602
Multicellular tumor spheroids (MCTS) are valuable in vitro tumor models frequently used to evaluate the penetration and efficacy of therapeutics. In this study, we evaluated potential differences in epigenetic markers, i.e., histone post-translational modifications (PTMs), in the layers of the HCT116 colon carcinoma MCTS. Cells were grown in agarose-coated 96 well plates, forming reproducible 1-mm-diameter MCTS. The MCTS were fractionated into three radially concentric portions, generating samples containing cells from the core, the mid and the external layers. Using mass spectrometry (MS)-based proteomics and EpiProfile, we quantified hundreds of histone peptides in different modified forms; by combining the results of all experiments, we quantified the abundance of 258 differently modified peptides, finding significant differences in their relative abundance across layers. Among these differences, we detected higher amounts of the repressive mark H3K27me3 in the external layers, compared to the core. We then evaluated the epigenetic response of MCTS following UNC1999 treatment, a drug targeting the enzymes that catalyze H3K27me3, namely, the polycomb repressive complex 2 (PRC2) subunits enhancer of zeste 1 (EZH1) and enhancer of zeste 2 (EZH2). UNC1999 treatment resulted in significant differences in MCTS diameter under drug treatment of varying duration. Using matrix-assisted laser desorption/ionization (MALDI) imaging, we determined that the drug penetrates the entire MCTS. Proteomic analysis revealed a decrease in abundance of H3K27me3, compared to the untreated sample, as expected. Interestingly, we observed a comparable growth curve for MCTS under constant drug treatment over 13 days with those treated for only 4 days at the beginning of their growth. We thus demonstrate that MS-based proteomics can define significant differences in histone PTM patterns in submillimetric layers of three-dimensional (3D) cultures. Moreover, we show that our model is suitable for monitoring drug localization and regulation of histone PTMs after drug treatment.
Co-reporter:Xiaoshan Yue, Jessica K. Lukowski, Eric M. Weaver, Susan B. Skube, and Amanda B. Hummon
Journal of Proteome Research 2016 Volume 15(Issue 12) pp:4265-4276
Publication Date(Web):October 3, 2016
DOI:10.1021/acs.jproteome.6b00342
Cell cultures are widely used model systems. Some immortalized cell lines can be grown in either two-dimensional (2D) adherent monolayers or in three-dimensional (3D) multicellular aggregates, or spheroids. Here, the quantitative proteome and phosphoproteome of colon carcinoma HT29 cells cultures in 2D monolayers and 3D spheroids were compared with a stable isotope labeling of amino acids (SILAC) labeling strategy. Two biological replicates from each sample were examined, and notable differences in both the proteome and the phosphoproteome were determined by nanoliquid chromatography tandem mass spectrometry (LC–MS/MS) to assess how growth configuration affects molecular expression. A total of 5867 protein groups, including 2523 phosphoprotein groups and 8733 phosphopeptides were identified in the samples. The Gene Ontology analysis revealed enriched GO terms in the 3D samples for RNA binding, nucleic acid binding, enzyme binding, cytoskeletal protein binding, and histone binding for their molecular functions (MF) and in the process of cell cycle, cytoskeleton organization, and DNA metabolic process for the biological process (BP). The KEGG pathway analysis indicated that 3D cultures are enriched for oxidative phosphorylation pathways, metabolic pathways, peroxisome pathways, and biosynthesis of amino acids. In contrast, analysis of the phosphoproteomes indicated that 3D cultures have decreased phosphorylation correlating with slower growth rates and lower cell-to-extracellular matrix interactions. In sum, these results provide quantitative assessments of the effects on the proteome and phosphoproteome of culturing cells in 2D versus 3D cell culture configurations.Keywords: 2D culture; 3D culture; colon cancer; phosphoproteomic; proteomic;
Co-reporter:Katelyn R. Ludwig; Richard Dahl
Journal of Proteome Research 2016 Volume 15(Issue 5) pp:1497-1505
Publication Date(Web):March 30, 2016
DOI:10.1021/acs.jproteome.5b01101
MicroRNAs (miRNAs) are post-transcriptional regulators of gene expression that are implicated in a number of disease states. MiRNAs can exist as individual entities or may be clustered and transcribed as a single polycistron. The mirn23a cluster consists of three miRNAs: miR-23a, miR-24-2, and miR-27a. Although these miRNAs are transcribed together, they often exist at varying levels in the cell. Despite the fact that the mirn23a cluster is known to play a role in a number of diseases and developmental processes, few direct targets have been identified. In this study, we examined the effects of miR-23a, miR-24-2, miR-27a, or the mirn23a cluster overexpression on the proteome of 70Z/3 pre-B lymphoblast cells. Quantitative mass spectrometry using isobaric tags for relative and absolute quantification (iTRAQ) allowed for the global profiling of cell lines after miRNA overexpression. We identified a number of targets of each miRNA that contained predicted miRNA seed sequences and are likely direct targets. In addition, we discovered a cohort of shared miRNA targets and cluster targets, demonstrating the importance of studying miRNA clusters in their entirety.
Co-reporter:Xiaoshan Yue, Alissa Schunter, and Amanda B. Hummon
Analytical Chemistry 2015 Volume 87(Issue 17) pp:8837
Publication Date(Web):August 3, 2015
DOI:10.1021/acs.analchem.5b01833
Phosphopeptide enrichment from complicated peptide mixtures is an essential step for mass spectrometry-based phosphoproteomic studies to reduce sample complexity and ionization suppression effects. Typical methods for enriching phosphopeptides include immobilized metal affinity chromatography (IMAC) or titanium dioxide (TiO2) beads, which have selective affinity and can interact with phosphopeptides. In this study, the IMAC enrichment method was compared with the TiO2 enrichment method, using a multistep enrichment strategy from whole cell lysate, to evaluate their abilities to enrich for different types of phosphopeptides. The peptide-to-beads ratios were optimized for both IMAC and TiO2 beads. Both IMAC and TiO2 enrichments were performed for three rounds to enable the maximum extraction of phosphopeptides from the whole cell lysates. The phosphopeptides that are unique to IMAC enrichment, unique to TiO2 enrichment, and identified with both IMAC and TiO2 enrichment were analyzed for their characteristics. Both IMAC and TiO2 enriched similar amounts of phosphopeptides with comparable enrichment efficiency. However, phosphopeptides that are unique to IMAC enrichment showed a higher percentage of multiphosphopeptides as well as a higher percentage of longer, basic, and hydrophilic phosphopeptides. Also, the IMAC and TiO2 procedures clearly enriched phosphopeptides with different motifs. Finally, further enriching with two rounds of TiO2 from the supernatant after IMAC enrichment or further enriching with two rounds of IMAC from the supernatant TiO2 enrichment does not fully recover the phosphopeptides that are not identified with the corresponding multistep enrichment.
Co-reporter:Katelyn R. Ludwig, Liangliang Sun, Guijie Zhu, Norman J. Dovichi, and Amanda B. Hummon
Analytical Chemistry 2015 Volume 87(Issue 19) pp:9532
Publication Date(Web):September 23, 2015
DOI:10.1021/acs.analchem.5b02457
Ultraperformance liquid chromatography (UPLC)-electrospray ionization (ESI)-tandem mass spectrometry (MS/MS) is typically employed for phosphoproteome analysis. Alternatively, capillary zone electrophoresis (CZE)-ESI-MS/MS has great potential for phosphoproteome analysis due to the significantly different migration times of phosphorylated and unphosphorylated forms of peptides. In this work, we systematically compared UPLC-MS/MS and CZE-MS/MS for phosphorylated peptide identifications (IDs) using an enriched phosphoproteome from the MCF-10A cell line. When the sample loading amount of UPLC was 10 times higher than that of CZE (2 μg vs 200 ng), UPLC generated more phosphorylated peptide IDs than CZE (3313 vs 1783). However, when the same sample loading amounts were used for CZE and UPLC (2–200 ng), CZE-MS/MS consistently and significantly outperformed UPLC-MS/MS in terms of phosphorylated peptide and total peptide IDs. This superior performance is most likely due to the higher peptide intensity generated by CZE-MS/MS. More importantly, compared with UPLC data from a 2 μg sample, CZE-MS/MS can identify over 500 unique phosphorylated peptides from a 200 ng sample, suggesting that CZE and UPLC are complementary for phosphorylated peptide IDs. With further improved loading capacity via a dynamic pH junction method, 2313 phosphorylated peptides were identified with single-shot CZE-MS/MS in a 100 min analysis. This number of phosphorylated peptide IDs is over 1 order of magnitude higher than the number of phosphorylated peptide IDs previously reported by single-shot CZE-MS/MS.
Co-reporter:Xin Liu and Amanda B. Hummon
Analytical Chemistry 2015 Volume 87(Issue 19) pp:9508
Publication Date(Web):June 18, 2015
DOI:10.1021/acs.analchem.5b00419
Co-reporter:Eric M. Weaver, Amanda B. Hummon and Richard B. Keithley
Analytical Methods 2015 vol. 7(Issue 17) pp:7208-7219
Publication Date(Web):24 Mar 2015
DOI:10.1039/C5AY00293A
As imaging mass spectrometry (IMS) has grown in popularity in recent years, the applications of this technique have become increasingly diverse. Currently there is a need for sophisticated data processing strategies that maximize the information gained from large IMS data sets. Traditional two-dimensional heat maps of single ions generated in IMS experiments lack analytical detail, yet manual analysis of multiple peaks across hundreds of pixels within an entire image is time-consuming, tedious and subjective. Here, various chemometric methods were used to analyze data sets obtained by matrix-assisted laser desorption/ionization (MALDI) IMS of multicellular spheroids. HT-29 colon carcinoma multicellular spheroids are an excellent in vitro model system that mimic the three dimensional morphology of tumors in vivo. These data are especially challenging to process because, while different microenvironments exist, the cells are clonal which can result in strong similarities in the mass spectral profiles within the image. In this proof-of-concept study, a combination of principal component analysis (PCA), clustering methods, and linear discriminant analysis was used to identify unique spectral features present in spatially heterogeneous locations within the image. Overall, the application of these exploratory data analysis tools allowed for the isolation and detection of proteomic changes within IMS data sets in an easy, rapid, and unsupervised manner. Furthermore, a simplified, non-mathematical theoretical introduction to the techniques is provided in addition to full command routines within the MATLAB programming environment, allowing others to easily utilize and adapt this approach.
Co-reporter:Xin Liu
Journal of The American Society for Mass Spectrometry 2015 Volume 26( Issue 4) pp:577-586
Publication Date(Web):2015 April
DOI:10.1007/s13361-014-1071-0
A new and simple method was developed to evaluate the distribution of therapeutics in three-dimensional multicellular tumor spheroids (MCTS) by combining serial trypsinization and nanoflow liquid chromatography-tandem mass spectrometry (nLC-MS/MS). This methodology was validated with quantitative measurements of irinotecan and its bioactive metabolite, SN-38, in distinct spatial regions of HCT 116 MCTS. Irinotecan showed a time-dependent permeability into MCTS with most of the drug accumulating in the core after 24 h of treatment. The amount of SN-38 detected was 30 times lower than that of the parent drug, and was more abundant in the outer rim and intermediate regions of MCTS where proliferating cells were present. This method can be used to investigate novel and established drugs. It enables investigation of drug penetration properties and identification of metabolites with spatial specificity in MCTS. The new approach has great value in facilitating the drug evaluation process.
Co-reporter:Kerry M. Bauer, Tanya N. Watts, Steven Buechler, and Amanda B. Hummon
Journal of Proteome Research 2014 Volume 13(Issue 11) pp:4910-4918
Publication Date(Web):2017-2-22
DOI:10.1021/pr500557n
Colon cancer is a major cause of cancer-related deaths worldwide. Adjuvant chemotherapy significantly reduces mortality in stage III colon cancer; however, it is only marginally effective in stage II patients. There is also increasing evidence that right-side colon cancer is different from left-side colon cancer. We have observed that the genes altered in expression between the poor and good prognosis tumors vary significantly depending on whether the malignancy originates on the right or left side of the colon. We have identified NADPH oxidase 4 (NOX4) to be highly predictive of relapse in stage II left-side colon cancer, whereas integrin alpha 3 beta 1 (ITGA3) is predictive of relapse in stage II right-side colon cancer. To investigate the underlying molecular mechanisms, we are analyzing the effect of ITGA3 and NOX4 silencing via RNA interference and pharmacological inhibition on global protein expression patterns via iTRAQ labeling and mass spectrometry in colon cancer cells. On the basis of bioinformatic analysis, the functions of these genes were assessed through phenotypic assays, revealing roles in cell migration and reactive oxygen species generation. These biomarkers for relapse risk are of clinical interest and lead to insight into how a tumor progresses to metastasis.
Co-reporter:Dorothy R. Ahlf, Rachel N. Masyuko, Amanda B. Hummon and Paul W. Bohn
Analyst 2014 vol. 139(Issue 18) pp:4578-4585
Publication Date(Web):24 Jun 2014
DOI:10.1039/C4AN00826J
A novel method of correlated imaging, combining confocal Raman microscopy (CRM) and matrix-assisted laser desorption ionization (MALDI) mass spectrometry imaging (MSI) was developed in order to investigate the structural and chemical diversity inherent in three-dimensional (3D) cell cultures. These 3D spheroidal cell cultures are high throughput in vitro model systems that recapitulate some of the chemical and physiological gradients characteristic of tissues. As a result, they are ideal for testing new imaging approaches due to the native diversity of cellular phenotypes found within a single culture. Individually, confocal Raman microscopy (CRM) and mass spectrometry imaging (MSI) produce different kinds of chemical information. CRM imaging reveals differences in cellular integrity and protein secretion across a typical near-equatorial transverse slice, while MSI shows localization of small molecules to discrete regions of the spheroid section. Correlating information obtained from these disparate imaging methods begins with an external fiducial mask, added to the spheroidal samples to orient image acquisition on the two orthogonal platforms. Rather than combine the images directly, principal component analysis is used to reveal the most chemically-informative elements, which are then combined using digital image correlation. Using this approach, relationships between the principal components of each method are visualized so that they may be compared on commensurate spatial length scales.
Co-reporter:Xin Liu, Eric M. Weaver, and Amanda B. Hummon
Analytical Chemistry 2013 Volume 85(Issue 13) pp:6295
Publication Date(Web):May 31, 2013
DOI:10.1021/ac400519c
Drug penetration into solid tumors is critical for the effectiveness of clinical chemotherapy. Failing to consider the efficiency of drug penetration can lead to fatal recurrence in many cancers. Three-dimensional (3D) cell cultures have served as an important model system and have contributed to valuable assays in drug discovery studies. However, limited methodologies result in incomplete evaluation of the distribution of many anticancer drugs. As a proof-of-concept study, we have applied matrix-assisted laser desorption/ionization (MALDI) imaging mass spectrometry (IMS) in HCT 116 colon carcinoma multicellular spheroids to assess the distribution of the anticancer drug, irinotecan. The time-dependent penetration of irinotecan was visualized and the localization of three metabolites as well as the parent drug in treated spheroids was mapped. To validate the identities of the metabolites, we analyzed extracts from drug-treated spheroids using nanoflow liquid chromatography-tandem mass spectrometry (nLC-MS/MS). Ten metabolites were identified with nLC-MS/MS, including those detected by MALDI IMS. This novel approach allows the measurement of drug penetration and distribution in 3D culture mimics and provides a more cost and time-effective approach for the testing of new pharmaceuticals compared to animal models.
Co-reporter:Leigh A. Weston, Kerry M. Bauer, Susan B. Skube, and Amanda B. Hummon
Analytical Chemistry 2013 Volume 85(Issue 22) pp:10675
Publication Date(Web):October 11, 2013
DOI:10.1021/ac401825m
Peptides are important species for a variety of biological functions. Detection and analysis of these molecules can be complicated by the presence of background matrix or contaminants. Therefore, a selective method to capture peptides could provide researchers with an option to isolate these remarkable species. Our goal was to perform a set of experiments that would validate the concept of a novel, selective peptide capture, whereby peptides are isolated on functionalized magnetic beads through the use of the heterobifunctional cross-linker, Sulfo-LC-SPDP. Matrix assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) was used to monitor mass changes associated with the cross-linking reaction. MALDI-TOF MS was then used to monitor conjugation between the cross-linked peptides and sulfhydryl magnetic beads by analyzing supernatant solutions for the presence or absence of cross-linked peptide. Through these experiments, we have proof of concept data confirming that peptides can be isolated on sulfhydryl magnetic beads by using Sulfo-LC-SPDP. This method is a suitable selective global peptide isolation strategy to separate the molecules from contaminating species or sample matrix. This novel method has a variety of potential applications and detection methods.
Co-reporter:Leigh A. Weston and Amanda B. Hummon
Analyst 2013 vol. 138(Issue 21) pp:6380-6384
Publication Date(Web):13 Sep 2013
DOI:10.1039/C3AN01121F
Primary tissue samples are valuable resources for investigators interested in understanding disease. In order to maximize the information content that can be gained from these precious samples, proper storage, handling, and preparation are essential. Some tissue preservation techniques utilize the cryopreservation medium, optimal cutting temperature (OCT) compound. While this medium provides benefits for traditional molecular studies, certain components can interfere with mass spectrometric analyses. Mass spectrometry based proteomics is a growing field with many applications for disease research. Our goal is to determine a reliable method for separating the proteins from the contaminating species in OCT embedded samples, thus making these samples compatible with mass spectrometric analyses. The novel applications of ether–methanol precipitation, filter-aided sample preparation (FASP), and SDS-PAGE provide researchers with protocols for removing OCT contaminating species from valuable samples. The results presented in this study show that all three methods reproducibly remove OCT; however, precipitation and FASP outperform SDS-PAGE by common proteomic metrics.
Co-reporter:Leigh A. Weston, Kerry M. Bauer and Amanda B. Hummon
Analytical Methods 2013 vol. 5(Issue 18) pp:4615-4621
Publication Date(Web):11 Jul 2013
DOI:10.1039/C3AY40853A
Discovery-based proteomic studies aim to answer important biological questions by identifying as many proteins as possible. In order to accomplish this lofty goal, an effort must be placed on determining an optimal workflow that maximizes protein identifications. In this study, we compare protein extraction, digestion and fractionation methods for bottom-up proteomics using a human colon cancer cell line as our model system. Four different buffers for protein extraction, two digestion approaches, as well as three sample fractionation methods were evaluated in order to determine an accessible workflow that gives maximal protein identifications. Samples comparing these workflows were analyzed via UPLC paired with tandem MS on a Q-Exactive mass spectrometer. Our goal is to determine an optimal workflow to enable users to maximize protein identifications. Our results show that an increased number of confident protein identifications are attained with a filter-aided digestion approach as compared to an in-solution digestion. Overall SDS-PAGE fractionation leads to higher numbers of identifications than SCX SpinTip and reverse phased cartridge platforms. The novel aspect of this work is the comparison of two readily available, offline platforms for fractionation in reference to a traditional technique, SDS-PAGE.
Co-reporter:Kerry M. Bauer and Amanda B. Hummon
Journal of Proteome Research 2012 Volume 11(Issue 9) pp:4744-4754
Publication Date(Web):2017-2-22
DOI:10.1021/pr300600r
The miR-143/-145 cluster is greatly reduced in several cancers, including colon cancer. Both miR-143 and miR-145 have been shown to possess antitumorigenic activity with involvement in various cancer-related events such as proliferation, invasion, and migration. As the deregulation of the miR-143/-145 cluster is implicated in tumorigenesis, we combined SILAC and microarray analyses to systematically interrogate the impact of miR-143/-145 on the colon cancer proteome and transcriptome. Using SILAC, we identified over 2000 proteins after reintroduction of miR-143 and miR-145, in the colon cancer cell line SW480, individually, and then, in concert. Our goal was to determine whether these microRNAs function individually or synergistically. The resulting regulated gene products showed evidence of both mRNA destabilization and translational inhibition with a bias toward the former mechanism of regulation. Numerous candidate targets were identified whose expression is attributable to an individual microRNA or whose regulation was more apparent following reintroduction of the miR-143/-145 cluster. In addition, several shared targets of miR-143 and miR-145 were identified. Overall, our results indicate that the summed effects of individually introduced microRNAs produce distinct molecular changes from the consequences of the assembled cluster. We conclude that there is a need to investigate both the individual and combined functional implications of a microRNA cluster.
Co-reporter:Xiao-Shan Yue
Frontiers in Biology 2012 Volume 7( Issue 6) pp:566-586
Publication Date(Web):2012 December
DOI:10.1007/s11515-012-2022-4
Phosphorylation is one of the most common post translational modifications (PTM), participating in a large number of processes to regulate cellular functions. Phosphorylation is also one of the key factors in the origin and development of cancer. The rapid development of mass spectrometric-based phosphoproteomic technologies has made it possible for high-throughput identification and quantification of phosphorylation events. In this review, we provide a general introduction and summary of the achievements made in mass spectrometry based phosphoproteomic research, including the approaches for phosphopeptide identification and quantification, as well as instrumentation and data interpretation methods. We also review some discoveries in cancer research made possible by phosphoproteomic analysis technologies.
Co-reporter:Haohang Li and Amanda B. Hummon
Analytical Chemistry 2011 Volume 83(Issue 22) pp:8794
Publication Date(Web):October 12, 2011
DOI:10.1021/ac202356g
Three-dimensional (3D) cell cultures have increased complexity compared to simple monolayer and suspension cultures, recapitulating the cellular architecture and molecular gradients in tissue. As such, they are popular for in vitro models in biological research. Classical imaging methodologies, like immunohistochemistry, are commonly used to examine the distribution of specific species within the spheroids. However, there is a need for an unbiased discovery-based methodology that would allow examination of protein/peptide distributions in 3D culture systems, without a need for prior knowledge of the analytes. We have developed a matrix-assisted laser desorption/ionization-mass spectrometry (MALDI-MS)-based imaging approach to examine protein distributions in 3D cell culture models. Using colon carcinoma cell lines, we detect changes in the spatial distribution of proteins across 3D culture structures. To identify the protein species present, we are combining results from the MS/MS capabilities of MALDI-MS to sequence peptides in a de novo fashion and nanoflow liquid chromatography–tandem mass spectrometry (nLC–MS/MS) of homogenized cultures. As a proof-of-principle, we have identified cytochrome C and Histone H4 as two of the predominant protein species in the 3D colon carcinoma cultures.
Co-reporter:Monica M. Schroll, Xin Liu, Sarah K. Herzog, Susan B. Skube, Amanda B. Hummon
Nutrition Research (October 2016) Volume 36(Issue 10) pp:1068-1080
Publication Date(Web):1 October 2016
DOI:10.1016/j.nutres.2016.08.002
Nutrient restriction, also known as caloric restriction, has been extensively examined for its positive impact on lifespan, immune system boost, and aging. In addition, nutrient restriction is implicated in decreasing cancer initiation and progression. Given the phenotypic changes associated with nutrient restriction, we hypothesized significant protein expression alterations must be associated with caloric restriction. To compare the molecular and phenotypic changes caused by glucose restriction and fetal bovine serum restriction there is need for an efficient model system. We establish 3-dimensional cell culture models, known as spheroids, in the HCT 116 colorectal cancer cell line as a high throughput model for studying the proteomic changes associated with nutrient restriction. Flow cytometry was used to assess apoptosis and autophagy levels in the spheroids under nutrient restriction. Isobaric tags for relative and absolute quantification and liquid chromatography tandem mass spectrometry were used to determine differential protein abundances between the nutrient restriction conditions. We identified specific proteins that have implications in cancer progression and metastasis that are differentially regulated by restriction of either glucose or serum. These proteins include the up-regulation of sirtuin 1 and protein inhibitor of activated STAT 1 and down-regulation of multi-drug resistance protein and Zinc finger and BTB domain-containing protein 7A. The results indicate nutrient restriction causes lower apoptotic and higher autophagy rates in HCT 116 spheroids. In addition, proteins shown to be differentially regulated by both glucose and serum restriction were similarly regulated.Download high-res image (132KB)Download full-size image
Co-reporter:Eric M. Weaver, Amanda B. Hummon
Advanced Drug Delivery Reviews (July 2013) Volume 65(Issue 8) pp:1039-1055
Publication Date(Web):1 July 2013
DOI:10.1016/j.addr.2013.03.006
Imaging mass spectrometry (IMS) has been a useful tool for investigating protein, peptide, drug and metabolite distributions in human and animal tissue samples for almost 15 years. The major advantages of this method include a broad mass range, the ability to detect multiple analytes in a single experiment without the use of labels and the preservation of biologically relevant spatial information. Currently the majority of IMS experiments are based on imaging animal tissue sections or small tumor biopsies. An alternative method currently being developed is the application of IMS to three-dimensional cell and tissue culture systems. With new advances in tissue culture and engineering, these model systems are able to provide increasingly accurate, high-throughput and cost-effective models that recapitulate important characteristics of cell and tissue growth in vivo. This review will describe the most recent advances in IMS technology and the bright future of applying IMS to the field of three-dimensional cell and tissue culture.Download high-res image (181KB)Download full-size image
Co-reporter:
Analytical Methods (2009-Present) 2015 - vol. 7(Issue 17) pp:NaN7219-7219
Publication Date(Web):2015/03/24
DOI:10.1039/C5AY00293A
As imaging mass spectrometry (IMS) has grown in popularity in recent years, the applications of this technique have become increasingly diverse. Currently there is a need for sophisticated data processing strategies that maximize the information gained from large IMS data sets. Traditional two-dimensional heat maps of single ions generated in IMS experiments lack analytical detail, yet manual analysis of multiple peaks across hundreds of pixels within an entire image is time-consuming, tedious and subjective. Here, various chemometric methods were used to analyze data sets obtained by matrix-assisted laser desorption/ionization (MALDI) IMS of multicellular spheroids. HT-29 colon carcinoma multicellular spheroids are an excellent in vitro model system that mimic the three dimensional morphology of tumors in vivo. These data are especially challenging to process because, while different microenvironments exist, the cells are clonal which can result in strong similarities in the mass spectral profiles within the image. In this proof-of-concept study, a combination of principal component analysis (PCA), clustering methods, and linear discriminant analysis was used to identify unique spectral features present in spatially heterogeneous locations within the image. Overall, the application of these exploratory data analysis tools allowed for the isolation and detection of proteomic changes within IMS data sets in an easy, rapid, and unsupervised manner. Furthermore, a simplified, non-mathematical theoretical introduction to the techniques is provided in addition to full command routines within the MATLAB programming environment, allowing others to easily utilize and adapt this approach.
Co-reporter:
Analytical Methods (2009-Present) 2013 - vol. 5(Issue 18) pp:
Publication Date(Web):
DOI:10.1039/C3AY40853A
Discovery-based proteomic studies aim to answer important biological questions by identifying as many proteins as possible. In order to accomplish this lofty goal, an effort must be placed on determining an optimal workflow that maximizes protein identifications. In this study, we compare protein extraction, digestion and fractionation methods for bottom-up proteomics using a human colon cancer cell line as our model system. Four different buffers for protein extraction, two digestion approaches, as well as three sample fractionation methods were evaluated in order to determine an accessible workflow that gives maximal protein identifications. Samples comparing these workflows were analyzed via UPLC paired with tandem MS on a Q-Exactive mass spectrometer. Our goal is to determine an optimal workflow to enable users to maximize protein identifications. Our results show that an increased number of confident protein identifications are attained with a filter-aided digestion approach as compared to an in-solution digestion. Overall SDS-PAGE fractionation leads to higher numbers of identifications than SCX SpinTip and reverse phased cartridge platforms. The novel aspect of this work is the comparison of two readily available, offline platforms for fractionation in reference to a traditional technique, SDS-PAGE.
.jpg)





















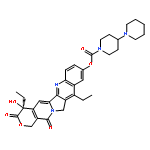
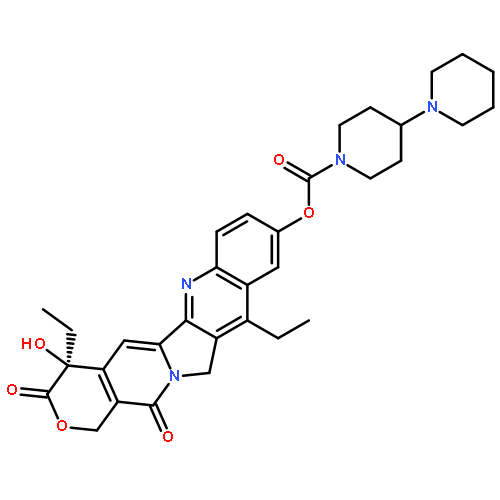
![1-Piperidinecarboxylicacid, 4-amino-,(4S)-4,11-diethyl-3,4,12,14-tetrahydro-4-hydroxy-3,14-dioxo-1H-pyrano[3',4':6,7]indolizino[1,2-b]quinolin-9-ylester](http://img.cochemist.com/ccimg/185400/185304-42-1.png)
![1-Piperidinecarboxylicacid, 4-amino-,(4S)-4,11-diethyl-3,4,12,14-tetrahydro-4-hydroxy-3,14-dioxo-1H-pyrano[3',4':6,7]indolizino[1,2-b]quinolin-9-ylester](http://img.cochemist.com/ccimg/185400/185304-42-1_b.png)
![1-Piperidinecarboxylicacid, 4-[(4-carboxybutyl)amino]-,(4S)-4,11-diethyl-3,4,12,14-tetrahydro-4-hydroxy-3,14-dioxo-1H-pyrano[3',4':6,7]indolizino[1,2-b]quinolin-9-ylester](http://img.cochemist.com/ccimg/181500/181467-56-1.png)
![1-Piperidinecarboxylicacid, 4-[(4-carboxybutyl)amino]-,(4S)-4,11-diethyl-3,4,12,14-tetrahydro-4-hydroxy-3,14-dioxo-1H-pyrano[3',4':6,7]indolizino[1,2-b]quinolin-9-ylester](http://img.cochemist.com/ccimg/181500/181467-56-1_b.png)
![Sulfo-N-succinimidyl 6-[3-(2-Pyridyldithio)propionamido] Hexanoate, Sodium Salt](http://img.cochemist.com/ccimg/169800/169751-10-4.png)
![Sulfo-N-succinimidyl 6-[3-(2-Pyridyldithio)propionamido] Hexanoate, Sodium Salt](http://img.cochemist.com/ccimg/169800/169751-10-4_b.png)
![Borate(1-),[5-[(3,5-dimethyl-2H-pyrrol-2-ylidene-kN)methyl]-1H-pyrrole-2-propanoato(2-)-kN1]difluoro-, hydrogen (1:1),(T-4)-](/data/chemimg/109100/165599-63-3.png)
![Borate(1-),[5-[(3,5-dimethyl-2H-pyrrol-2-ylidene-kN)methyl]-1H-pyrrole-2-propanoato(2-)-kN1]difluoro-, hydrogen (1:1),(T-4)-](/data/chemimg/109100/165599-63-3_b.png)




![b-D-Glucopyranosiduronic acid,(4S)-4,11-diethyl-3,4,12,14-tetrahydro-4-hydroxy-3,14-dioxo-1H-pyrano[3',4':6,7]indolizino[1,2-b]quinolin-9-yl](http://img.cochemist.com/ccimg/121100/121080-63-5.png)
![b-D-Glucopyranosiduronic acid,(4S)-4,11-diethyl-3,4,12,14-tetrahydro-4-hydroxy-3,14-dioxo-1H-pyrano[3',4':6,7]indolizino[1,2-b]quinolin-9-yl](http://img.cochemist.com/ccimg/121100/121080-63-5_b.png)