Co-reporter:Andrew M. Camp, Matthew R. Kita, Javier Grajeda, Peter S. White, Diane A. Dickie, and Alexander J. M. Miller
Inorganic Chemistry September 18, 2017 Volume 56(Issue 18) pp:11141-11141
Publication Date(Web):September 5, 2017
DOI:10.1021/acs.inorgchem.7b01485
A protocol for identifying ligand binding modes in a series of iridium pincer complexes bearing hemilabile aza-crown ether ligands has been developed using readily accessible NMR methods. The approach was tested on a collection of 13 structurally diverse pincer–crown ether complexes that include several newly characterized species. New synthetic routes enable facile interconversion of coordination modes and supporting ligands. Detailed structural assignments of five complexes reveal that the difference in chemical shift (Δδ) between geminal protons in the crown ether is influenced by diamagnetic anisotropy arising from halides and other ligands in the primary coordination sphere. The average difference in chemical shift between diastereotopic geminal protons in the crown ether macrocycle (Δδavg), as determined through a single 1H–13C HSQC experiment, provides information on the pincer ligand binding mode by establishing whether the macrocycle is in close proximity to the metal center. The Δδavg values for binding modes that involve chelating ether(s) bound to iridium are roughly 2-fold larger than those for tridentate complexes with no Ir–O bonds.
Co-reporter:Catherine L. Pitman and Alexander J. M. Miller
Organometallics May 22, 2017 Volume 36(Issue 10) pp:1906-1906
Publication Date(Web):May 2, 2017
DOI:10.1021/acs.organomet.7b00175
An iridium methyl complex, [Cp*Ir(bpy)(CH3)]+, was prepared by electrophilic methylation of Cp*Ir(bpy) with CH3I and characterized electrochemically, photophysically, crystallographically, and computationally. Irradiation of the MLCT transition of [Cp*Ir(bpy)(CH3)]+ in the presence of CH3I in acetonitrile produces ethane, methane, propionitrile, and succinonitrile. A series of mechanistic studies indicates that C–C bond formation is mediated by free methyl radicals produced through monometallic photochemical homolysis of the Ir–CH3 bond.
Co-reporter:Brian M. Lindley, Quinton J. Bruch, Peter S. White, Faraj Hasanayn, and Alexander J. M. Miller
Journal of the American Chemical Society April 19, 2017 Volume 139(Issue 15) pp:5305-5305
Publication Date(Web):April 6, 2017
DOI:10.1021/jacs.7b01323
The conversion of metal nitride complexes to ammonia may be essential to dinitrogen fixation. We report a new reduction pathway that utilizes ligating acids and metal–ligand cooperation to effect this conversion without external reductants. Weak acids such as 4-methoxybenzoic acid and 2-pyridone react with nitride complex [(H-PNP)RuN]+ (H-PNP = HN(CH2CH2PtBu2)2) to generate octahedral ammine complexes that are κ2-chelated by the conjugate base. Experimental and computational mechanistic studies reveal the important role of Lewis basic sites proximal to the acidic proton in facilitating protonation of the nitride. The subsequent reduction to ammonia is enabled by intramolecular 2H+/2e– proton-coupled electron transfer from the saturated pincer ligand backbone.
Co-reporter:Brian M. Lindley, Quinton J. Bruch, Peter S. White, Faraj Hasanayn, and Alexander J. M. Miller
Journal of the American Chemical Society April 19, 2017 Volume 139(Issue 15) pp:5305-5305
Publication Date(Web):April 6, 2017
DOI:10.1021/jacs.7b01323
The conversion of metal nitride complexes to ammonia may be essential to dinitrogen fixation. We report a new reduction pathway that utilizes ligating acids and metal–ligand cooperation to effect this conversion without external reductants. Weak acids such as 4-methoxybenzoic acid and 2-pyridone react with nitride complex [(H-PNP)RuN]+ (H-PNP = HN(CH2CH2PtBu2)2) to generate octahedral ammine complexes that are κ2-chelated by the conjugate base. Experimental and computational mechanistic studies reveal the important role of Lewis basic sites proximal to the acidic proton in facilitating protonation of the nitride. The subsequent reduction to ammonia is enabled by intramolecular 2H+/2e– proton-coupled electron transfer from the saturated pincer ligand backbone.
Co-reporter:Eric S. Wiedner, Matthew B. Chambers, Catherine L. Pitman, R. Morris Bullock, Alexander J. M. Miller, and Aaron M. Appel
Chemical Reviews 2016 Volume 116(Issue 15) pp:8655-8692
Publication Date(Web):August 2, 2016
DOI:10.1021/acs.chemrev.6b00168
Transition metal hydrides play a critical role in stoichiometric and catalytic transformations. Knowledge of free energies for cleaving metal hydride bonds enables the prediction of chemical reactivity, such as for the bond-forming and bond-breaking events that occur in a catalytic reaction. Thermodynamic hydricity is the free energy required to cleave an M–H bond to generate a hydride ion (H–). Three primary methods have been developed for hydricity determination: the hydride transfer method establishes hydride transfer equilibrium with a hydride donor/acceptor pair of known hydricity, the H2 heterolysis method involves measuring the equilibrium of heterolytic cleavage of H2 in the presence of a base, and the potential–pKa method considers stepwise transfer of a proton and two electrons to give a net hydride transfer. Using these methods, over 100 thermodynamic hydricity values for transition metal hydrides have been determined in acetonitrile or water. In acetonitrile, the hydricity of metal hydrides spans a range of more than 50 kcal/mol. Methods for using hydricity values to predict chemical reactivity are also discussed, including organic transformations, the reduction of CO2, and the production and oxidation of hydrogen.
Co-reporter:Catherine L. Pitman; Kelsey R. Brereton
Journal of the American Chemical Society 2016 Volume 138(Issue 7) pp:2252-2260
Publication Date(Web):January 17, 2016
DOI:10.1021/jacs.5b12363
Aqueous hydride transfer is a fundamental step in emerging alternative energy transformations such as H2 evolution and CO2 reduction. “Hydricity,” the hydride donor ability of a species, is a key metric for understanding transition metal hydride reactivity, but comprehensive studies of aqueous hydricity are scarce. An extensive and self-consistent aqueous hydricity scale is constructed for a family of Ru and Ir hydrides that are key intermediates in aqueous catalysis. A reference hydricity is determined using redox potentiometry and spectrophotometric titration for a particularly water-soluble species. Then, relative hydricity values for a range of species are measured using hydride transfer equilibria, taking advantage of expedient new synthetic procedures for Ru and Ir hydrides. This large collection of hydricity values provides the most comprehensive picture so far of how ligands impact hydricity in water. Strikingly, we also find that hydricity can be viewed as a continuum in water: the free energy of hydride transfer changes with pH, buffer composition, and salts present in solution.
Co-reporter:Matthew B. Chambers, Daniel A. Kurtz, Catherine L. Pitman, M. Kyle Brennaman, and Alexander J. M. Miller
Journal of the American Chemical Society 2016 Volume 138(Issue 41) pp:13509-13512
Publication Date(Web):September 27, 2016
DOI:10.1021/jacs.6b08701
Artificial photosynthesis relies on coupling light absorption with chemical fuel generation. A mechanistic study of visible light-driven H2 production from [Cp*Ir(bpy)H]+ (1) has revealed a new, highly efficient pathway for integrating light absorption with bond formation. The net reaction of 1 with a proton source produces H2, but the rate of excited state quenching is surprisingly acid-independent and displays no observable deuterium kinetic isotopic effect. Time-resolved photoluminescence and labeling studies are consistent with diffusion-limited bimetallic self-quenching by electron transfer. Accordingly, the quantum yield of H2 release nearly reaches unity as the concentration of 1 increases. This unique pathway for photochemical H2 generation provides insight into transformations catalyzed by 1.
Co-reporter:C. L. Pitman, O. N. L. Finster and A. J. M. Miller
Chemical Communications 2016 vol. 52(Issue 58) pp:9105-9108
Publication Date(Web):01 Mar 2016
DOI:10.1039/C6CC00575F
Attempts to generate a proposed rhodium hydride catalytic intermediate instead resulted in isolation of (Cp*H)Rh(bpy)Cl (1), a pentamethylcyclopentadiene complex, formed by C–H bond-forming reductive elimination from the fleeting rhodium hydride. The hydride transfer ability of diene 1 was explored through thermochemistry and hydride transfer reactions, including the reduction of NAD+.
Co-reporter:Kelsey R. Brereton, Catherine L. Pitman, Thomas R. Cundari, and Alexander J. M. Miller
Inorganic Chemistry 2016 Volume 55(Issue 22) pp:12042
Publication Date(Web):November 9, 2016
DOI:10.1021/acs.inorgchem.6b02223
The hydricity of the heterobimetallic iridium/ruthenium catalyst [Cp*Ir(H)(μ-bpm)Ru(bpy)2]3+ (1, where Cp* = η5-pentamethylcyclopentadienyl, bpm = 2,2′-bipyrimidine, and bpy = 2,2′-bipyridine) has been determined in both acetonitrile (63.1 kcal mol–1) and water (29.7 kcal mol–1). Hydride 1 features a large increase in the hydride donor ability when the solvent is changed from acetonitrile to water. The acidity of 1, in contrast, is essentially solvent-independent because 1 remains strongly acidic in both solvents. On the basis of an X-ray crystallographic study, spectroscopic analysis, and time-dependent density functional theory calculations, the disparate reactivity trends are ascribed to substantial delocalization of the electron density onto both the bpm and bpy ligands in the conjugate base of 1, [Cp*Ir(μ-bpm)Ru(bpy)2]2+ (3). The H2 evolution tendencies of 1 are considered in the context of thermodynamic parameters.
Co-reporter:A. G. Walden, A. Kumar, N. Lease, A. S. Goldman and A. J. M. Miller
Dalton Transactions 2016 vol. 45(Issue 24) pp:9766-9769
Publication Date(Web):16 Mar 2016
DOI:10.1039/C6DT00522E
With a view towards replacing sacrificial hydrogen acceptors in alkane dehydrogenation catalysis, electrochemical methods for oxidative activation of a pincer-ligated iridium hydride intermediate were explored. A 1H+/2e− oxidation process was observed in THF solvent, with net hydride loss leading to a reactive cationic intermediate that can be trapped by chloride. Analogous reactivity was observed with the concerted hydride transfer reagent Ph3C+, connecting chemical and electrochemical hydride loss pathways.
Co-reporter:Simon J. Meek; Catherine L. Pitman
Journal of Chemical Education 2016 Volume 93(Issue 2) pp:275-286
Publication Date(Web):January 19, 2016
DOI:10.1021/acs.jchemed.5b00160
An introductory guide to deducing the mechanism of chemical reactions is presented. Following a typical workflow for probing reaction mechanism, the guide introduces a wide range of kinetic and mechanistic tools. In addition to serving as a broad introduction to mechanistic analysis for students and researchers, the guide has also been used by instructors to provide the organizational structure for an upper-level course on organic and inorganic reaction mechanism. After providing students with the tools of mechanistic study, student-led discussions of case studies and an independent proposal project provide preparation for understanding the mechanism of new reactions encountered in independent research.
Co-reporter:Kelsey R. BreretonSarina M. Bellows, Hengameh Fallah, Antonio A. Lopez, Robert M. Adams, Alexander J. M. Miller, William D. JonesThomas R. Cundari
The Journal of Physical Chemistry B 2016 Volume 120(Issue 50) pp:12911-12919
Publication Date(Web):December 8, 2016
DOI:10.1021/acs.jpcb.6b09864
Hydricity, or hydride donating ability, is a thermodynamic value that helps define the reactivity of transition metal hydrides. To avoid some of the challenges of experimental hydricity measurements in water, a computational method for the determination of aqueous hydricity values has been developed. With a thermochemical cycle involving deprotonation of the metal hydride (pKa), 2e– oxidation of the metal (E°), and 2e– reduction of the proton, hydricity values are provided along with other valuable thermodynamic information. The impact of empirical corrections (for example, calibrating reduction potentials with 2e– organic versus 1e– inorganic potentials) was assessed in the calculation of the reduction potentials, acidities, and hydricities of a series of iridium hydride complexes. Calculated hydricities are consistent with electronic trends and agree well with experimental values.
Co-reporter:Brian M. Lindley, Aaron M. Appel, Karsten Krogh-Jespersen, James M. Mayer, and Alexander J. M. Miller
ACS Energy Letters 2016 Volume 1(Issue 4) pp:698
Publication Date(Web):September 9, 2016
DOI:10.1021/acsenergylett.6b00319
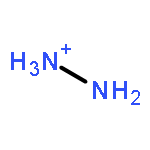
The development of a sustainable ammonia synthesis by proton-coupled electroreduction of dinitrogen (N2) requires knowledge of the thermodynamics described by standard reduction potentials. The first collection of N2 reduction standard potentials in an organic solvent are reported here. The potentials for reduction of N2 to ammonia (NH3), hydrazine (N2H4), and diazene (N2H2) in acetonitrile (MeCN) solution are derived using thermochemical cycles. Ammonia is thermodynamically favored, with a 0.43 V difference between NH3 and N2H4 and a 1.26 V difference between NH3 and N2H2. The thermodynamics for reduction of N2 to the protonated products ammonium (NH4+) and hydrazinium (N2H5+) under acidic conditions are also presented. Comparison with the H+/H2 potential in MeCN reveals a 63 mV thermodynamic preference for N2 reduction to NH3 over H2 production. Combined with knowledge of the kinetics of electrode-catalyzed H2 evolution, a wide working region is identified to guide future electrocatalytic studies.
Co-reporter:Javier Grajeda, Matthew R. Kita, Lauren C. Gregor, Peter S. White, and Alexander J. M. Miller
Organometallics 2016 Volume 35(Issue 3) pp:306-316
Publication Date(Web):November 19, 2015
DOI:10.1021/acs.organomet.5b00786
Several new iridium(I) and iridium(III) carbonyl complexes supported by aminophosphinite pincer ligands have been prepared and characterized. A surprising diversity of reaction pathways was encountered upon treatment of Ir carbonyl complexes with Li+, Na+, Ca2+, and La3+ salts. Iridium(III) hydridocarbonyl chloride complexes underwent either halide abstraction or halide substitution reactions, whereas iridium(I) carbonyl complexes underwent protonative oxidative addition reactions. When the nitrogen donor of the pincer ligand is an aza-crown ether macrocycle, cation–macrocycle interactions could be supported, leading to divergent reactivity in some cases.
Co-reporter:Lauren C. Gregor, Javier Grajeda, Matthew R. Kita, Peter S. White, Andrew J. Vetter, and Alexander J. M. Miller
Organometallics 2016 Volume 35(Issue 17) pp:3074-3086
Publication Date(Web):August 30, 2016
DOI:10.1021/acs.organomet.6b00607
The rate of catalytic methanol carbonylation to acetic acid is typically limited by either the oxidative addition of methyl iodide or the subsequent C–C bond-forming migratory insertion step. These elementary steps have been studied independently in acetonitrile solution for iridium aminophenylphosphinite (NCOP) complexes. The modular synthesis of NCOP ligands containing a macrocyclic aza-crown ether arm enables a direct comparison of two complementary catalyst optimization strategies: synthetic modification of the phenyl backbone and noncovalent modification through cation–crown interactions with Lewis acids in the surrounding environment. The oxidative addition of methyl iodide to iridium(I) carbonyl complexes proceeds readily at room temperature to form iridium(III) methylcarbonyliodide complexes. The methyl complexes undergo migratory insertion under 1 atm CO at 70 °C to produce iridium(III) acetyl species. Synthetic tuning, by incorporation of a methoxy group into the ligand backbone, had little influence on the rate. The addition of lithium and lanthanum salts, in contrast, enhanced the rate of C–C bond formation up to 25-fold. In the case of neutral iodide complexes, mechanistic studies suggest that Lewis acidic cations act as halide abstractors. In halide-free, cationic iridium complexes, the cations bind the macrocyclic ligand arm, further activating the iridium(III) center. The macrocyclic ligand is essential to the observed reactivity: complexes supported by acyclic diethylamine-containing ligands underwent migratory insertion slowly, Lewis acid effects were negligible, and the acetyl products decomposed over time.
Co-reporter:Andrew G. Walden and Alexander J. M. Miller
Chemical Science 2015 vol. 6(Issue 4) pp:2405-2410
Publication Date(Web):04 Feb 2015
DOI:10.1039/C5SC00032G
The tris(2-pyridyl)phosphine oxide (Py3PO) complex [Ru(Py3PO)(bpy)(OH2)]2+ (bpy is 2,2′-bipyridine) is a pH-dependent water oxidation electrocatalyst that accelerates dramatically with increasing pH—up to 780 s−1 at pH 10 (∼1 V overpotential). Despite retaining the pentakis(pyridine) ligand arrangement common to previously reported catalysts, the tripodal Py3PO ligand framework supports much faster electrocatalysis. The early stages of the catalytic cycle are proposed to follow the typical pattern of single-site ruthenium catalysts, with two sequential 1H+/1e− proton-coupled electron transfer (PCET) oxidations, but the pH-dependent onset of catalysis and rapid rates are distinguishing features of the present system.
Co-reporter:Seth M. Barrett, Samuel A. Slattery, and Alexander J. M. Miller
ACS Catalysis 2015 Volume 5(Issue 11) pp:6320
Publication Date(Web):September 28, 2015
DOI:10.1021/acscatal.5b01995
The mechanism of photochemical formic acid dehydrogenation catalyzed by [Cp*Ir(bpy)(Cl)]+ (1, bpy = 2,2′-bipyridine) and [Cp*Ir(bpy-OMe)(Cl)]+ (1-OMe, bpy-OMe = 4,4′-dimethoxy-2,2′-bipyridine) is examined. The catalysts operate with good turnover frequency (TOF) across an unusually wide pH range. Above pH 7, the evolved gas is >95% pure H2 (along with traces of CO2 but no detectable CO). Light-triggered H2 release from a metal hydride intermediate is found to be the turnover-limiting step, based on the observed first-order dependence on catalyst concentration, saturation behavior in formate concentration, and direct in situ observation of a metal hydride resting state during turnover. Deactivation pathways are identified, including ligand loss and aggregate formation, precipitation of insoluble forms of the catalyst, and deprotonation of the iridium hydride intermediate. Guided by mechanistic insights, improved catalytic activity (initial TOF exceeding 50 h–1), stability (>500 turnovers at nearly 5 atm), and selectivity (>95% H2 gas) are achieved.Keywords: hydride; hydrogen evolution; hydrogen storage; iridium; photocatalysis
Co-reporter:Jacob B. Smith and Alexander J. M. Miller
Organometallics 2015 Volume 34(Issue 19) pp:4669-4677
Publication Date(Web):July 7, 2015
DOI:10.1021/acs.organomet.5b00405
Nickel catalysts supported by diethylamine- or aza-crown ether-containing aminophosphinite (NCOP) pincer ligands catalyze the insertion of benzaldehyde into a C–H bond of acetonitrile. The catalytic activity of neutral (NCOP)Ni(OtBu) and cationic [(NCOP)Ni(NCCH3)]+ are starkly different. The neutral tert-butoxide precatalysts are active without any added base and give good yields of product after 24 h, while the cationic precatalysts require a base cocatalyst and still operate much more slowly (120 h in typical runs). A series of in situ spectroscopic studies identified several intermediates, including a nickel cyanoalkoxide complex that was observed in all of the reactions regardless of the choice of precatalyst. Reaction monitoring also revealed that the neutral tert-butoxide precatalysts decompose to form the cationic acetonitrile complex during catalysis; this deactivation involves alkoxide abstraction and can be hastened by the addition of lithium salts. While the deactivated cationic species is inactive under standard base-free conditions, catalysis can be reinitiated by the addition of catalytic amounts of base.
Co-reporter:Matthew R. Kita
Journal of the American Chemical Society 2014 Volume 136(Issue 41) pp:14519-14529
Publication Date(Web):October 2, 2014
DOI:10.1021/ja507324s
Complexes of a new multidentate ligand combining a rigid, strongly donating pincer scaffold with a flexible, weakly donating aza-crown ether moiety are reported. The pincer-crown ether ligand exhibits tridentate, tetradentate, and pentadentate coordination modes. The coordination mode can be changed by Lewis base displacement of the chelating ethers, with binding equilibria dramatically altered through lithium and sodium cation–macrocycle interactions. Cation-promoted hydrogen activation was accomplished by an iridium monohydride cation ligated in a pentadentate fashion by the pincer-crown ether ligand. The rate can be controlled on the basis of the choice of cation (with lithium-containing reactions proceeding about 10 times faster than sodium-containing reactions) or on the basis of the concentration of the cation. Up to 250-fold rate enhancements in H/D exchange rates are observed when catalytic amounts of Li+ are added.
Co-reporter:Seth M. Barrett ; Catherine L. Pitman ; Andrew G. Walden
Journal of the American Chemical Society 2014 Volume 136(Issue 42) pp:14718-14721
Publication Date(Web):October 9, 2014
DOI:10.1021/ja508762g
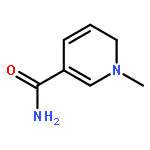
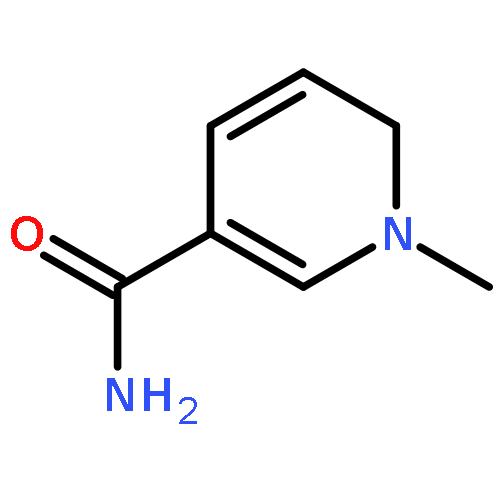
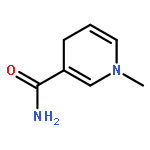
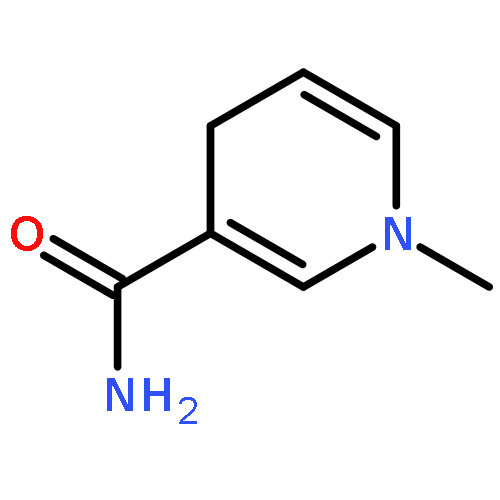
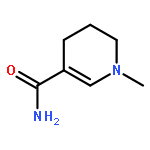
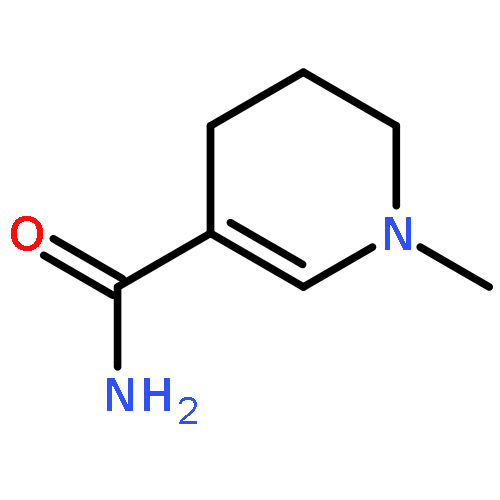
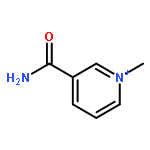
Visible light-triggered hydride transfer from [Cp*Ir(bpy)(H)]+ (1) to organic acids and 1-methylnicotinamide (MNA+) is reported (Cp* = pentamethylcyclopentadienyl; bpy = 2,2′-bipyridine). A new thermochemical cycle for determining excited-state hydride donor ability (hydricity) predicted that 1 would be an incredibly potent photohydride in acetonitrile. Phototriggered H2 release was indeed observed from 1 in the presence of various organic acids, providing experimental evidence for an increase in hydricity of at least 18 kcal/mol in the excited state. The rate and product selectivity of hydride transfer to MNA+ are photoswitchable: 1,4-dihydro-1-methylnicotinamide forms slowly in the dark but rapidly under illumination, and photolysis can also produce doubly reduced 1,4,5,6-tetrahydro-1-methylnicotinamide.
Co-reporter:Catherine L. Pitman and Alexander J. M. Miller
ACS Catalysis 2014 Volume 4(Issue 8) pp:2727
Publication Date(Web):July 7, 2014
DOI:10.1021/cs500441f
A light-activated hydrogen evolution electrocatalyst is reported. Hydrogen evolves near the thermodynamic potential when aqueous solutions of the iridium chloride complex [Cp*Ir(bpy)(Cl)][Cl] (1, bpy = 2,2′-bipyridine) are illuminated by visible light. In the dark, no electrocatalytic activity is observed. This unique hydrogen evolution mechanism is made possible because a single transition metal complex is the active light absorber and active electrocatalyst. Optimization by tuning the electronic structure of the catalyst and varying reaction conditions resulted in H2 evolution with faster rates, even at milder applied potentials (kobs ∼ 0.1 s–1 at 100 mV electrochemical overpotential).Keywords: electrocatalysis; hydride; hydrogen evolution; iridium; photocatalysis; photoelectrocatalysis
Co-reporter:Alexander J. M. Miller, Werner Kaminsky, and Karen I. Goldberg
Organometallics 2014 Volume 33(Issue 5) pp:1245-1252
Publication Date(Web):February 20, 2014
DOI:10.1021/om5000166
An arene activation strategy for the selective disassembly of aryl ethers is reported. A variety of aryl ethers readily bind an electrophilic pentamethylcyclopentadienyl iridium center by η6-arene coordination, generating complexes that are activated toward hydrolysis and cleavage of the Ar–OR bond (R = Me, Et, Ph). Hydrolysis occurs rapidly at room temperature in aqueous pH 7 phosphate buffer (or upon modest heating under acidic conditions), releasing alcohol while forming cyclohexadienyl-one products. Under strongly acidic conditions, protonation of the dienyl-one followed by substitution with starting aryl ether completes a hydrolysis cycle. Mechanistic studies suggest that the key hydrolysis step proceeds via nucleophilic attack at the ipso position of the arene (SNAr mechanism). The observed mechanism is considered in the context of lignocellulosic biomass conversion.
Co-reporter:Timothy P. Brewster ; Alexander J. M. Miller ; D. Michael Heinekey ;Karen I. Goldberg
Journal of the American Chemical Society 2013 Volume 135(Issue 43) pp:16022-16025
Publication Date(Web):October 21, 2013
DOI:10.1021/ja408149n
A series of half-sandwich Ir and Rh compounds are demonstrated to be competent catalysts for the hydrogenation of carboxylic acids under relatively mild conditions. Of the structurally diverse group of catalysts tested for activity, a Cp*Ir complex supported by an electron-releasing 2,2′-bipyridine ligand was the most active. Higher activity was achieved with employment of Brønsted or Lewis acid promoters. Mechanistic studies suggest a possible reaction pathway involving activated carboxylic acid substrates. The hydrogenation reaction was shown to be general to a variety of aliphatic acids.
Co-reporter: Alexer J. M. Miller; D. Michael Heinekey; James M. Mayer; Karen I. Goldberg
Angewandte Chemie 2013 Volume 125( Issue 14) pp:4073-4076
Publication Date(Web):
DOI:10.1002/ange.201208470
Co-reporter: Alexer J. M. Miller; D. Michael Heinekey; James M. Mayer; Karen I. Goldberg
Angewandte Chemie International Edition 2013 Volume 52( Issue 14) pp:3981-3984
Publication Date(Web):
DOI:10.1002/anie.201208470
Co-reporter:Andrew G. Walden and Alexander J. M. Miller
Chemical Science (2010-Present) 2015 - vol. 6(Issue 4) pp:NaN2410-2410
Publication Date(Web):2015/02/04
DOI:10.1039/C5SC00032G
The tris(2-pyridyl)phosphine oxide (Py3PO) complex [Ru(Py3PO)(bpy)(OH2)]2+ (bpy is 2,2′-bipyridine) is a pH-dependent water oxidation electrocatalyst that accelerates dramatically with increasing pH—up to 780 s−1 at pH 10 (∼1 V overpotential). Despite retaining the pentakis(pyridine) ligand arrangement common to previously reported catalysts, the tripodal Py3PO ligand framework supports much faster electrocatalysis. The early stages of the catalytic cycle are proposed to follow the typical pattern of single-site ruthenium catalysts, with two sequential 1H+/1e− proton-coupled electron transfer (PCET) oxidations, but the pH-dependent onset of catalysis and rapid rates are distinguishing features of the present system.
Co-reporter:A. G. Walden, A. Kumar, N. Lease, A. S. Goldman and A. J. M. Miller
Dalton Transactions 2016 - vol. 45(Issue 24) pp:NaN9769-9769
Publication Date(Web):2016/03/16
DOI:10.1039/C6DT00522E
With a view towards replacing sacrificial hydrogen acceptors in alkane dehydrogenation catalysis, electrochemical methods for oxidative activation of a pincer-ligated iridium hydride intermediate were explored. A 1H+/2e− oxidation process was observed in THF solvent, with net hydride loss leading to a reactive cationic intermediate that can be trapped by chloride. Analogous reactivity was observed with the concerted hydride transfer reagent Ph3C+, connecting chemical and electrochemical hydride loss pathways.
Co-reporter:C. L. Pitman, O. N. L. Finster and A. J. M. Miller
Chemical Communications 2016 - vol. 52(Issue 58) pp:NaN9108-9108
Publication Date(Web):2016/03/01
DOI:10.1039/C6CC00575F
Attempts to generate a proposed rhodium hydride catalytic intermediate instead resulted in isolation of (Cp*H)Rh(bpy)Cl (1), a pentamethylcyclopentadiene complex, formed by C–H bond-forming reductive elimination from the fleeting rhodium hydride. The hydride transfer ability of diene 1 was explored through thermochemistry and hydride transfer reactions, including the reduction of NAD+.
.jpg)