Co-reporter:Peter B. Karadakov, David L. Cooper
Computational and Theoretical Chemistry 2017 Volume 1116(Volume 1116) pp:
Publication Date(Web):15 September 2017
DOI:10.1016/j.comptc.2017.01.003
•Five 10-π-electron fused conjugated systems with cyclopropenyl rings are planar (MP2).•Spin-coupled calculations provide familiar patterns of well-localized active orbitals.•Aromatic ground states: in-phase combination of two “’Kekulé” Rumer structures.•First singlet electronic excited states probably antiaromatic: “antiresonance”.The feasibilities and electronic structures of five ten-π-electron fused conjugated molecules involving cyclopropenyl rings are explored using second-order Møller-Plesset perturbation theory (MP2), spin-coupled (SC) and complete-active-space self-consistent-field (CASSCF) wavefunctions, in the cc-pVTZ basis. All five fused conjugated molecules are predicted to have rigid planar ground state geometries of C2v or D2h symmetry and large dipole moments (if not of D2h symmetry). The compact ground state SC(10) wavefunctions with ten active orbitals for these molecules are found to be of comparable quality to the respective CASSCF(10,10) constructions, but much easier to interpret. The analyses of the ground state SC(10) wavefunctions for all five fused conjugated molecules reveal resonance patterns which indicate that all of these molecules are aromatic in their electronic ground states; on the other hand, the SC(10) approximations to the first singlet electronic excited states are found to exhibit “antiresonance” which suggests that each of the five molecules switches from aromatic to antiaromatic upon vertical excitation from the ground state to its first singlet excited state. Ring strain prevents the formation of a fused structure involving three cyclopropenyl rings and a cycloheptatrienyl ring; the alternative stable dehydro compound which resembles m-benzyne is shown, using a SC(12) wavefunction, to involve a weak σ bond between the dehydro centres.Download high-res image (206KB)Download full-size image
Co-reporter:David L. Cooper, Fabio E. Penotti, Robert Ponec
Computational and Theoretical Chemistry 2017 Volume 1116(Volume 1116) pp:
Publication Date(Web):15 September 2017
DOI:10.1016/j.comptc.2016.12.010
•Domain-averaged Fermi hole analysis of CASSCF(18,14): 3-center 4-electron π bonding.•Localized natural orbitals from CASSCF(18,14) and icMRCI support DAFH picture of O3.•Defined new (improved) three-center bonding index Y(A, B, C).•Rival VB constructions assessed using CASSCF(18,14) and icMRCI π-only Y(O1, O2, O3).•Fixed-orbital SCc(10)⊕SCd(10)⊕SCI(10) combination performs well.Domain-averaged Fermi hole analysis is carried out for the ground state of O3 at its equilibrium geometry using a complete-active-space self-consistent field CASSCF(18,14) wavefunction, based on a slightly expanded full-valence active space. This initial analysis is augmented with an examination of the corresponding localized natural orbitals (LNOs) and of the numerical values obtained with a new improved definition of three-center bond indices for correlated singlet systems. Much the same pattern of LNOs is observed when using instead a subsequent internally-contracted multiconfiguration-reference configuration interaction construction, which also provides very similar values for the three-center bond indices. This gives us confidence to use such bond indices, alongside relative energies and the electric dipole moment, to assess the relative merits of various combinations of spin-coupled (full generalized valence bond) components with ten active electrons: four π, four σ bonding and the two nonbonding σ electrons associated with the central O atom. These multi-component valence bond descriptions were generated either with or without subsequent orbital reoptimization. The description of the π system which emerges from all of our analysis conforms to a standard model of three-center four-electron π bonding that incorporates a nontrivial degree of (partial) diradical character. Whereas certain combinations of ten-electron spin-coupled components can faithfully reproduce such a picture, none of the individual rival components appears to have sufficient flexibility on its own.Download high-res image (155KB)Download full-size image
Co-reporter:Peter B. Karadakov and David L. Cooper
The Journal of Physical Chemistry A 2016 Volume 120(Issue 43) pp:8769-8779
Publication Date(Web):October 14, 2016
DOI:10.1021/acs.jpca.6b09426
Spin-coupled (SC) theory is used to obtain modern valence-bond descriptions of the electronic structures of local minimum and transition-state geometries of three species that have been considered to exhibit homoconjugation and homoaromaticity: the homotropenylium ion, C8H9+, the cycloheptatriene neutral ring, C7H8, and the 1,3-bishomotropenylium ion, C9H11+. The resulting compact SC wave functions are of comparable quality to complete-active-space self-consistent field constructions that are based on the same “N electrons in M orbitals” active spaces, but they are much easier to interpret directly. Analysis of the forms of the SC orbitals and of the overlaps between them, as well as an examination of the compositions of the associated resonance patterns, strongly suggest that both of the homotropenylium and 1,3-bishomotropenylium ions are homoaromatic at their local minimum geometries, with all of the other cases that were considered being nonaromatic. The SC results also show that the differences between “no-bond” and “bond” homoconjugated systems are very likely to be much smaller than previously thought.
Co-reporter:Željka Krpetić, Adam M. Davidson, Martin Volk, Raphaël Lévy, Mathias Brust, and David L. Cooper
ACS Nano 2013 Volume 7(Issue 10) pp:8881
Publication Date(Web):September 23, 2013
DOI:10.1021/nn403350v
Differential centrifugal sedimentation (DCS) has been applied to accurately size ligand-protected gold hydrosols in the 10 to 50 nm range. A simple protocol is presented to correct for particle density variations due to the presence of the ligand shell, which is formed here by either polyethylene glycol-substituted alkane thiols (PEG-alkane thiols) of different chain length or oligopeptides. The method gives reliable data for all particle sizes investigated and lends itself to rapid routine sizing of nanoparticles. Unlike TEM, DCS is highly sensitive to small changes in the thickness of the organic ligand shell and can be applied to monitor shell thickness variations of as little as 0.1 nm on particles of a given core size.Keywords: differential centrifugal sedimentation (DCS); FTIR; gold hydrosols; monolayer-protected clusters (MPCs); nanoparticles; particle sizing; peptide-capped nanoparticles
Co-reporter:Jabir H.A. Al-Fahemi, David L. Cooper, Neil L. Allan
Journal of Molecular Structure: THEOCHEM 2009 Volume 901(1–3) pp:56-59
Publication Date(Web):15 May 2009
DOI:10.1016/j.theochem.2009.01.007
Momentum-space descriptors are used to construct compact multiple linear regression models for the toxicity to Tetrahymena pyriformis of two sets of molecules examined previously (A.G. Saliner, X. Gironés, Theochem 727 (2005) 97). The resulting few-descriptor models for 30 aliphatic molecules and for 48 anilines compare favorably with the earlier study of these two sets of molecules. Furthermore, models based on a very small number of trivial feature count descriptors (numbers of atoms of a given type and the number of bonds) are found to be of practically the same statistical quality as those that require quantum mechanical calculations.
Co-reporter:J. Grant Hill, David L. Cooper and Peter B. Karadakov
The Journal of Physical Chemistry A 2008 Volume 112(Issue 50) pp:12823-12828
Publication Date(Web):September 17, 2008
DOI:10.1021/jp800969k
The electronic rearrangements along the lowest-energy path for the gas-phase retro Diels−Alder reaction of norbornene are monitored using spin-coupled theory. We find that the most dramatic changes to the electronic structure occur in a relatively narrow interval in which the system passes through a geometry at which it can be considered to be significantly aromatic. We provide an estimate of the vertical resonance energy. Our results are consistent with the anticipated synchronous “aromatic” nature of this reaction, but we find that the key changes occur a little before the actual transition state is reached.








![Tricyclo[6.2.0.03,6]deca-1,3,5,7,9-pentaene](http://img.cochemist.com/ccimg/24500/24447-42-5.png)
![Tricyclo[6.2.0.03,6]deca-1,3,5,7,9-pentaene](http://img.cochemist.com/ccimg/24500/24447-42-5_b.png)
![Phosphoric acid, mono[2-(methylamino)ethyl] monooctadecyl ester](http://img.cochemist.com/ccimg/104800/104702-34-3.png)
![Phosphoric acid, mono[2-(methylamino)ethyl] monooctadecyl ester](http://img.cochemist.com/ccimg/104800/104702-34-3_b.png)








![Tricyclo[6.2.0.02,5]deca-1,3,5,7,9-pentaene](/data/chemimg/2719700/61960-82-5.png)
![Tricyclo[6.2.0.02,5]deca-1,3,5,7,9-pentaene](/data/chemimg/2719700/61960-82-5_b.png)















![Phosphoric acid, mono[2-[(1,1-dimethylethyl)amino]ethyl] monododecylester](http://img.cochemist.com/ccimg/139800/139715-71-2.png)
![Phosphoric acid, mono[2-[(1,1-dimethylethyl)amino]ethyl] monododecylester](http://img.cochemist.com/ccimg/139800/139715-71-2_b.png)












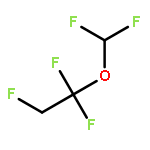
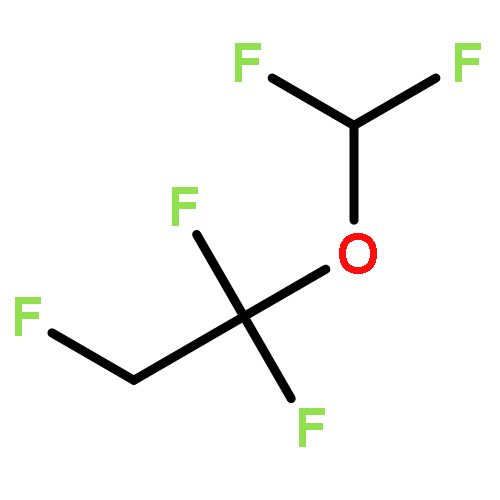

![Bicyclo[2.2.0]hexane](http://img.cochemist.com/ccimg/1600/1552-98-3.png)
![Bicyclo[2.2.0]hexane](http://img.cochemist.com/ccimg/1600/1552-98-3_b.png)




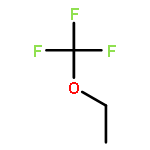
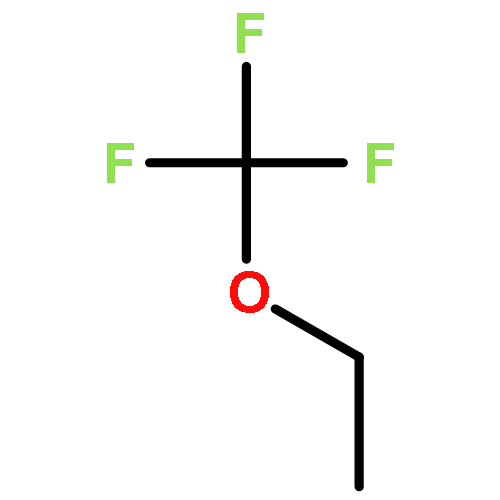




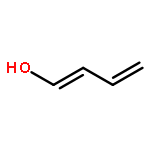
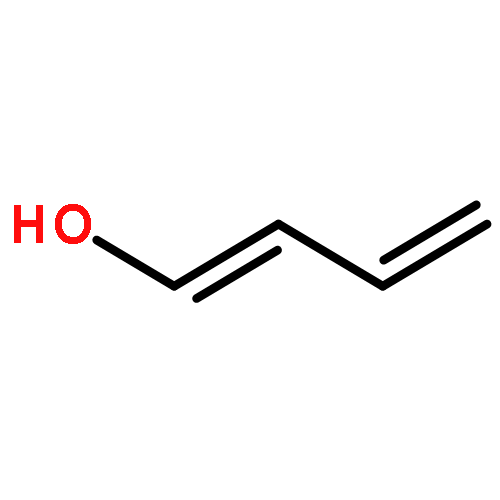
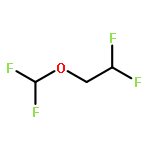



![Phosphoric acid, monododecyl mono[2-(methylamino)ethyl] ester](http://img.cochemist.com/ccimg/129300/129274-39-1.png)
![Phosphoric acid, monododecyl mono[2-(methylamino)ethyl] ester](http://img.cochemist.com/ccimg/129300/129274-39-1_b.png)


























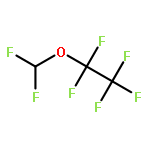
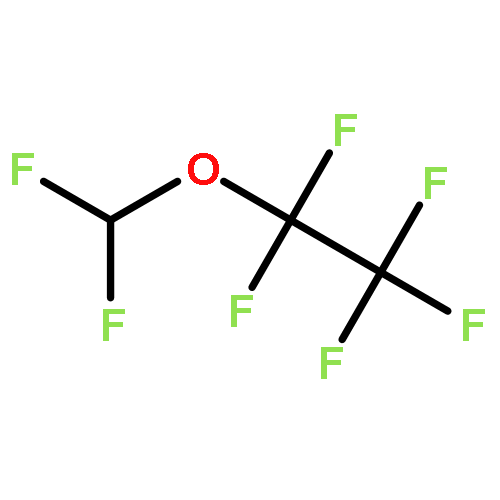

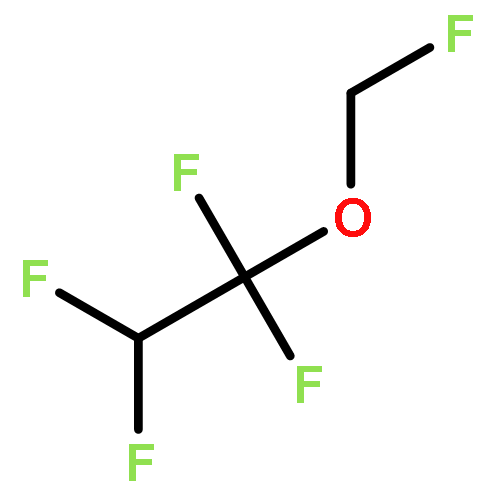





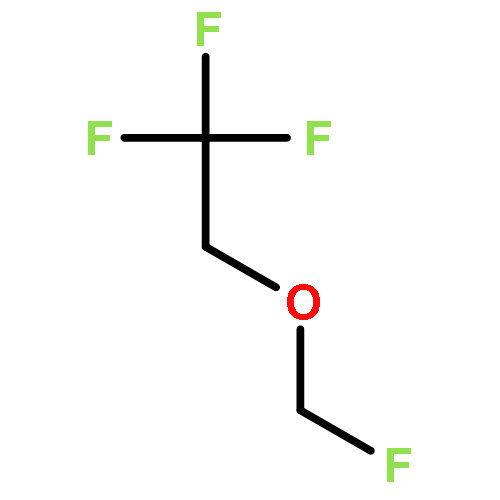










![Phosphoric acid,monohexyl mono[2-(methylamino)ethyl] ester](http://img.cochemist.com/ccimg/104800/104702-32-1.png)
![Phosphoric acid,monohexyl mono[2-(methylamino)ethyl] ester](http://img.cochemist.com/ccimg/104800/104702-32-1_b.png)