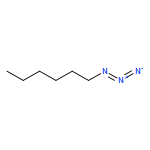
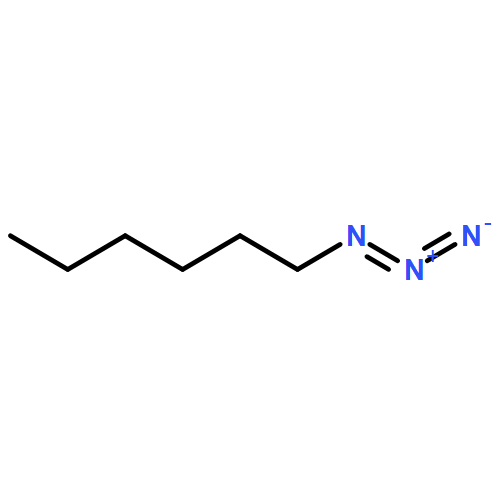
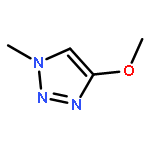
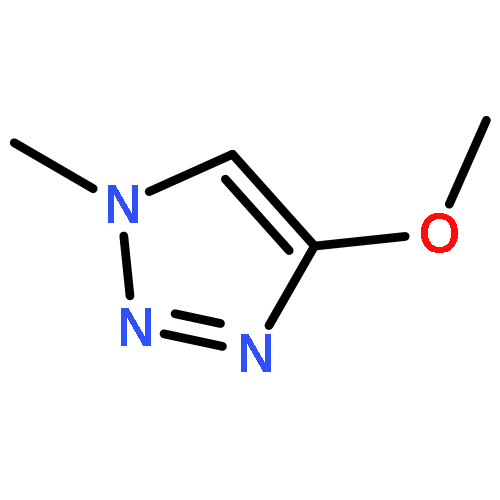
Co-reporter:Xavier Creary, Kyle Chormanski, Gabriel Peirats, and Carol Renneburg
The Journal of Organic Chemistry June 2, 2017 Volume 82(Issue 11) pp:5720-5720
Publication Date(Web):May 5, 2017
DOI:10.1021/acs.joc.7b00548
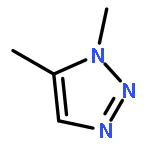
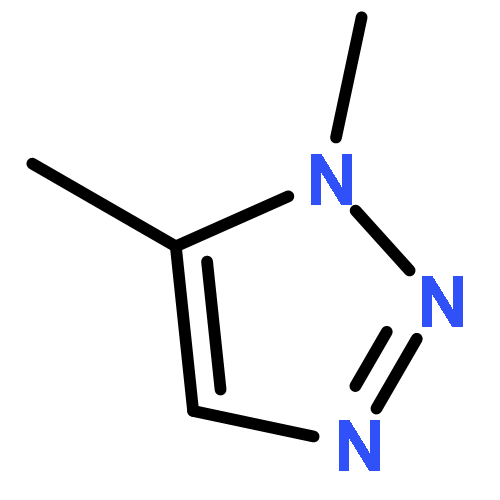
Three fluorobenzenes substituted with meta-triazole groups have been prepared, and 19F chemical shifts indicate that these triazole groups are all inductively electron-withdrawing in character, with the 1,5-triazole being the most electron-withdrawing. σ+ values for these three triazoles have also been determined from solvolysis rates of substituted cumyl trifluoroacetates. When substituted in the para-position, the 1,4 and the 2,4-triazoles are cation-stabilizing, whereas the 1,5-triazole is carbocation-destabilizing. γ+ values indicate that the 1,4 triazole group is cation-stabilizing relative to the phenyl group, albeit the 1,5 triazole is significantly destabilizing relative to phenyl. These studies all suggest that the 1,5-triazole group exerts a strong electron-withdrawing effect on carbocations that is not offset by a resonance effect. The three triazole groups all enhance the methylenecyclopropane rearrangement rate and are therefore radical stabilizers. The smallest stabilizing effect is seen for the 1,5-triazole, and this is attributed to the triazole group being twisted out of conjugation in the developing benzylic radical. Finally, the anionic triazole group is the most effective radical-stabilizing group. Computational studies indicate that these triazole groups all stabilize benzylic radicals by a spin delocalization mechanism.
Co-reporter:Xavier Creary
The Journal of Organic Chemistry 2015 Volume 80(Issue 22) pp:11378-11387
Publication Date(Web):October 26, 2015
DOI:10.1021/acs.joc.5b01955
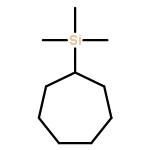
A series of γ-trimethylsilyl-substituted carbenes have been studied experimentally and by computational methods. In an acyclic system, 1,3-trimethylsilyl migration successfully competes with 1,3-hydrogen migration to the carbene center. The behavior of cyclic 3-trimethylsilyl-substituted carbenes contrasts with that of the acyclic system. Only 1,2-hydrogen migration processes are observed in the five-membered ring due to the high barrier to 1,3-hydrogen migration. In the cyclohexyl system, a small amount of a cyclopropane derived from 1,3-hydrogen migration occurs, as shown by a labeling study. In the cycloheptyl carbene system, a labeling study again showed that 1,3-hydrogen migration to the carbene center leads to the major product. Computational studies suggest that the cyclic carbenes all have lower energy conformations where the trimethylsilyl group is in a pseudo equatorial conformation where it cannot migrate to the carbene center. Computational studies also suggest that cyclohexyl and cycloheptyl carbene systems are slightly stabilized by a rear lobe interaction of the Si–C bond with the carbene center.
Co-reporter:Xavier Creary, Anna Heffron, Gabrielle Going, and Mariana Prado
The Journal of Organic Chemistry 2015 Volume 80(Issue 3) pp:1781-1788
Publication Date(Web):January 6, 2015
DOI:10.1021/jo502691t
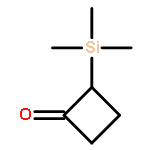
A series of isomeric 3-trimethylsilyl-1-arylcyclobutyl carbocations, 10 and 11, where the cross-ring 3-trimethylsilyl group has the potential to interact with the cationic center, have been generated under solvolytic conditions. When the cationic center can interact with the rear lobe of the carbon–silicon bond, rate enhancements become progressively larger as the substituent on the aryl group becomes more electron-withdrawing. When the potential interaction with the trimethylsilyl group is via a front lobe interaction, there is minimal rate enhancement over the range of substituents. Computational studies have also been carried out on these cations 10 and 11. Calculated trimethylsilyl stabilization energies progressively increase with electron-withdrawing character of the aryl groups when the trimethylsilyl interaction is via the rear lobe. By way of contrast, there are minimal changes in stabilization energies when the potential trimethylsilyl interaction is via the front lobe of the carbon–silicon bond. These computational studies, along with the solvolytic studies, point to a significant rear lobe 3-trimethylsilyl stabilization of arylcyclobutyl cations. They also argue against any front lobe stabilization of the isomeric arylcyclobutyl cations.
Co-reporter:Xavier Creary and Anna Heffron
The Journal of Organic Chemistry 2014 Volume 79(Issue 6) pp:2547-2555
Publication Date(Web):February 18, 2014
DOI:10.1021/jo500007p
endo-2-Trimethylsilyl-anti-7-norbornyl triflate undergoes solvolysis reactions 1.8 × 104 faster than 7-norbornyl triflate in CD3CO2D and 1.3 × 105 times faster in CF3CH2OH. The exclusive substitution products with retained stereochemistry point to a significantly stabilized γ-trimethylsilyl carbocation intermediate. The endo-2-trimethylsilyl-7-norbornyl carbene gives a major rearrangement product where the trimethylsilyl-activated hydrogen migrates to the carbenic center. This rearrangement product implies stabilization of the carbene by the γ-trimethylsilyl group. Isodesmic computational studies (M062X/6-311+G**) indicate that the endo-2-trimethylsilyl-7-norbornyl cation is stabilized by 16.2 kcal/mol and that the endo-2-trimethylsilyl-7-norbornyl carbene is stabilized by a smaller factor of 1.8 kcal/mol. By way of contrast, anti-7-trimethylsilyl-endo-2-norbornyl mesylate undergoes solvolysis in CD3CO2D only 2.6 times faster than endo-2-norbornyl mesylate and 9.4 times faster in CF3CH2OH. The substitution products have only partially retained stereochemistry, and significant rearrangements are observed. The anti-7-trimethylsilyl-2-norbornyl carbene gives a rearrangement product via 1,3-hydrogen migration of the C6 hydrogen, which is completely analogous to the behavior of the unsubstituted 2-norbornyl carbene. Isodesmic calculations show that the anti-7-trimethylsilyl-2-norbornyl cation is stabilized by only 3.2 kcal/mol relative to the 2-norbornyl cation, and the corresponding anti-7-trimethylsilyl-2-norbornyl carbene is stabilized by a negligible 0.9 kcal/mol.
Co-reporter:Xavier Creary
Journal of the American Chemical Society 2013 Volume 135(Issue 17) pp:6570-6578
Publication Date(Web):March 25, 2013
DOI:10.1021/ja400747u
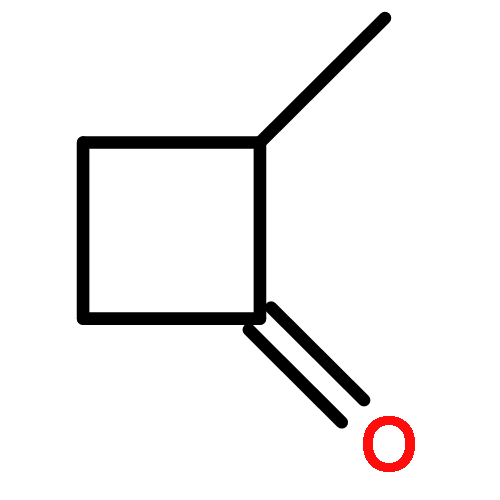
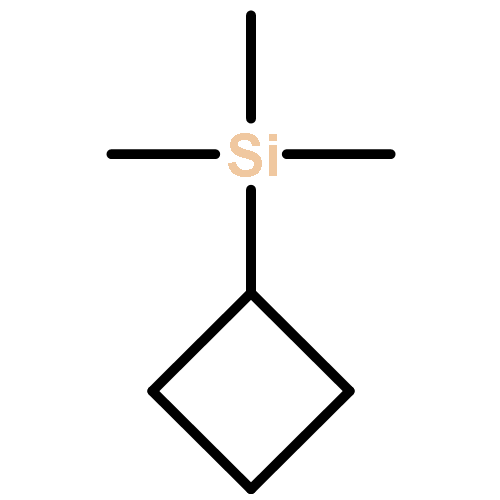
3-Trimethylsilylcyclobutylidene was generated by pyrolysis of the sodium salt of the tosylhydrazone derivative of 3-trimethylsilylcyclobutanone. This carbene converts to 1-trimethylsilylbicyclobutane as the major product. A labeling study shows that this intramolecular rearrangement product comes from 1,3-hydrogen migration to the carbenic center and not 1,3-silyl migration. Computational studies show two carbene minimum energy conformations, with the lower energy conformation displaying a large stabilizing interaction of the carbene center with the rear lobe of the C3–Si bond. In this conformation, the trimethylsilyl group cannot migrate to the carbene center, and the most favorable process is 1,3-hydrogen migration. When the carbene is generated photochemically in methanol, it reacts by a protonation mechanism giving the highly stabilized 3-trimethylsilylcyclobutyl carbocation as an intermediate. When generated in dimethylamine as solvent, the carbene undergoes preferred attack of this nucleophilic solvent from the back of this C–Si rear lobe stabilized carbene.
Co-reporter:Xavier Creary, Andrew Anderson, Carl Brophy, Frances Crowell, and Zachary Funk
The Journal of Organic Chemistry 2012 Volume 77(Issue 19) pp:8756-8761
Publication Date(Web):August 15, 2012
DOI:10.1021/jo301265t
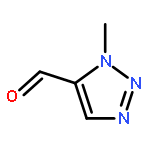
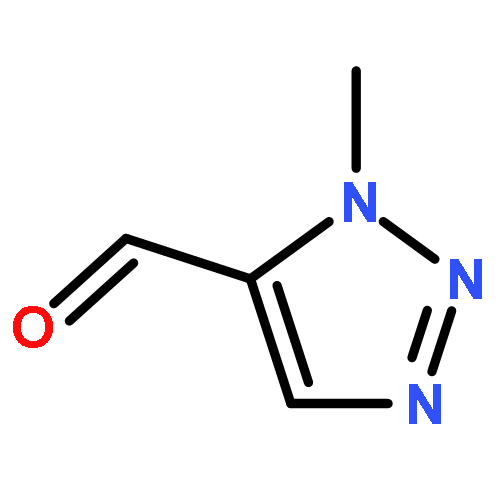
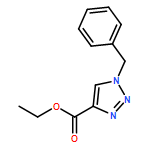
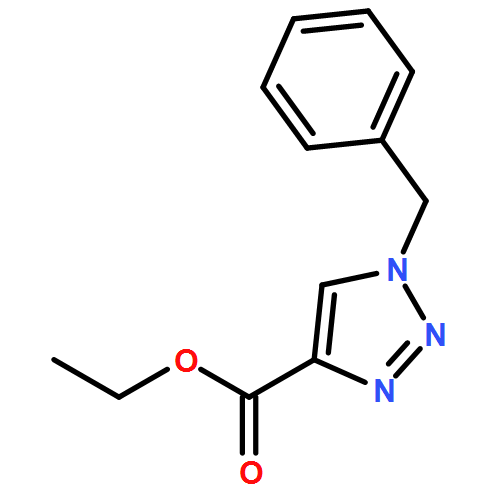
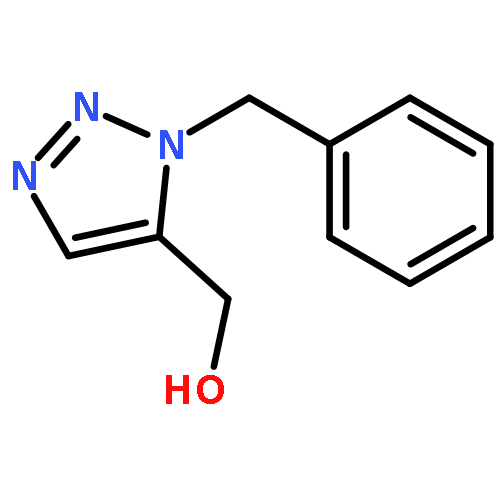
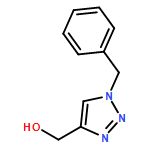
1,4-Disubstituted-1H-1,2,3-triazoles 1 can easily be distinguished from the isomeric 1,5-disubstituted-1H-1,2,3-triazoles 2 by simple one-dimensional 13C NMR spectroscopy using gated decoupling. The C5 signal of 1 appears at δ ∼120 ppm, while the C4 signal of 2 appears at δ ∼133 ppm. Computational studies also predict the upfield shift of C5 of 1 relative to C4 in 2.
Co-reporter:Xavier Creary, Jenifer Hinckley, Casey Kraft, and Madeleine Genereux
The Journal of Organic Chemistry 2011 Volume 76(Issue 7) pp:2062-2071
Publication Date(Web):March 3, 2011
DOI:10.1021/jo102309w
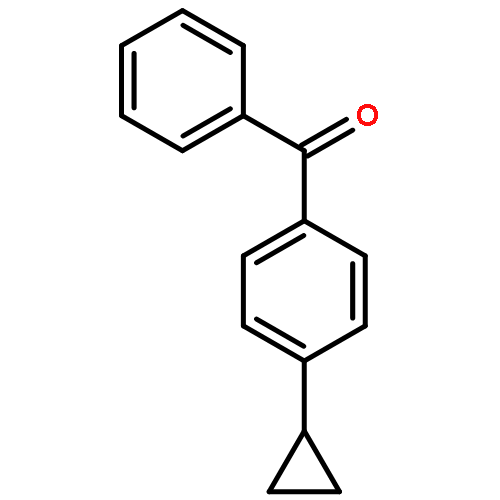
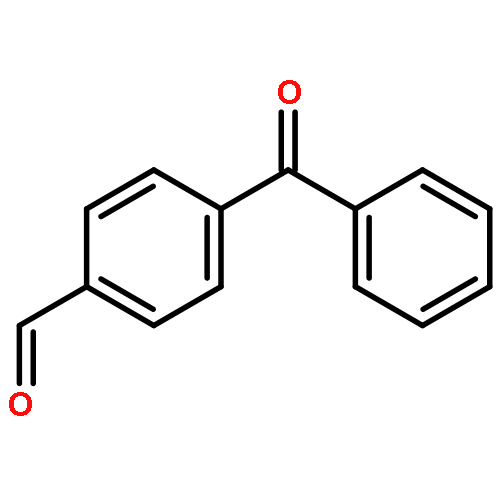
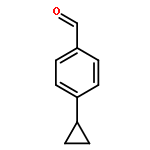
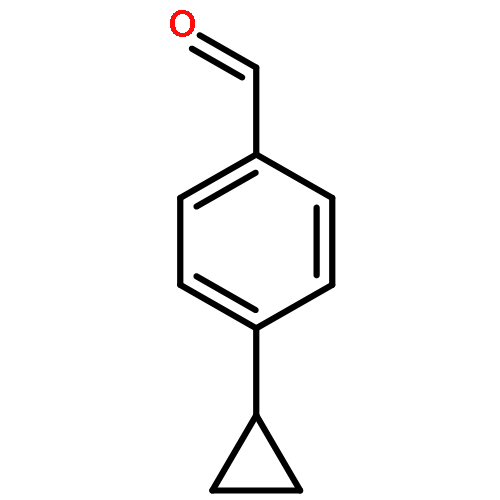
p-Cyclopropylbenzophenone, 20, gives no photoreduction when irradiated in i-PrOH solvent. This is a general phenomenon and a number of cyclopropyl-substituted benzophenones, including 4-(endo-6-bicyclo[3.1.0]hexyl)benzophenone, 19, 4-(cis-2,3-dimethylcyclopropyl)benzophenone, 21, 4-(cis-2-vinylcyclopropyl)benzophenone, 22, and 4-(endo-7-bicyclo[4.1.0]hept-2-enyl)benzophenone, 23, also fail to undergo photoreduction. Instead these latter compounds undergo cis−trans isomerization when irradiated. A mechanism involving formation of an (n, π*) triplet, which subsequently fragments the strained cyclopropane bond to give a lower energy and unreactive open triplet, has been suggested. p-Cyclopropylvalerophenone, 25, and p-(endo-6-bicyclo[3.1.0]hexyl)valerophenone, 24, also undergo photoisomerization and fail to undergo the Norrish Type II photoreactions. Triplet energy dissipation by fragmentation of the cyclopropane bond is also proposed. In addition to the Norrish Type II reaction, p-cyclobutylvalerophenone, 27, undergoes a photofragmentation to give ethylene and p-vinylvalerophenone, 60, by an energy dissipation mechanism involving a 1,4-biradical derived from cyclobutane bond fragmentation.
.jpg)
![SILANE, BICYCLO[4.1.0]HEPT-1-YLTRIMETHYL-](http://img.cochemist.com/ccimg/77600/77508-36-2.png)
![SILANE, BICYCLO[4.1.0]HEPT-1-YLTRIMETHYL-](http://img.cochemist.com/ccimg/77600/77508-36-2_b.png)


















































![Ruthenium,chloro[(1,2,3,4,5-h)-1,2,3,4,5-pentamethyl-2,4-cyclopentadien-1-yl]bis(triphenylphosphine)-](http://img.cochemist.com/ccimg/92400/92361-49-4.png)
![Ruthenium,chloro[(1,2,3,4,5-h)-1,2,3,4,5-pentamethyl-2,4-cyclopentadien-1-yl]bis(triphenylphosphine)-](http://img.cochemist.com/ccimg/92400/92361-49-4_b.png)
![Ethanone, 1-[1-(phenylmethyl)-1H-1,2,3-triazol-4-yl]-](/data/chemimg/1022700/80819-67-6.png)
![Ethanone, 1-[1-(phenylmethyl)-1H-1,2,3-triazol-4-yl]-](/data/chemimg/1022700/80819-67-6_b.png)



















![phenyl[4-(propan-2-yl)phenyl]methanone](http://img.cochemist.com/ccimg/18900/18864-76-1.png)
![phenyl[4-(propan-2-yl)phenyl]methanone](http://img.cochemist.com/ccimg/18900/18864-76-1_b.png)
![Silane, bicyclo[1.1.0]but-1-yltrimethyl-](http://img.cochemist.com/ccimg/17100/17049-34-2.png)
![Silane, bicyclo[1.1.0]but-1-yltrimethyl-](http://img.cochemist.com/ccimg/17100/17049-34-2_b.png)