Co-reporter:Chao Gao, Ting-Hong Ye, Cui-Ting Peng, Yao-jie Shi, Xin-Yu You, Lu Xiong, Kai Ran, Li-Dan Zhang, Xiu-Xiu Zeng, Ning-Yu Wang, Luo-Ting Yu, Yu-Quan Wei
Biomedicine & Pharmacotherapy 2017 Volume 88(Volume 88) pp:
Publication Date(Web):1 April 2017
DOI:10.1016/j.biopha.2017.01.098
New chemotherapeutic compounds and regimens are needed to combat multidrug-resistant Mycobacterium tuberculosis. Here, we used a series of murine models to assess an antitubercular lead compound SKLB-TB1001. In the Mycobacterium bovis bacillus Calmette-Guérin and the acute M. tuberculosis H37Rv infection mouse models, SKLB-TB1001 significantly attenuated the mycobacterial load in lungs and spleens. The colony forming unit counts and histological examination of lungs from H37Rv infected mice revealed that the benzothiazinethione analogue SKLB-TB1001 as a higher dose level was as effective as isoniazid. Moreover, in a multidrug-resistant (MDR)-TB mouse model, SKLB-TB1001 showed significant activity in a dose-dependent manner and was more effective than streptomycin. These results suggested that SKLB-TB1001 could be an antitubercular drug candidate worth further investigation.
Co-reporter:Zhihao Liu;Qian Lei;Wei Wei;Lu Xiong;Yaojie Shi;Guoyi Yan;Chao Gao;Tinghong Ye;Ningyu Wang;Luoting Yu
RSC Advances (2011-Present) 2017 vol. 7(Issue 44) pp:27737-27746
Publication Date(Web):2017/05/22
DOI:10.1039/C7RA02518A
Polo-like kinase 4 (PLK4), a vital regulator of centriole duplication, is important for maintaining genome stability. Dysregulation of PLK4 has been found in several human cancers and is associated with a predisposition to tumorigenesis. Herein, we describe the discovery of (E)-4-(3-arylvinyl-1H-indazol-6-yl)pyrimidin-2-amine derivatives as potent PLK4 inhibitors with more concise structure using a scaffold hopping strategy. SAR exploration and preliminary assessment identified 14i as a new PLK4 inhibitor which displayed excellent potency in vitro. 14i could inhibit the activity of PLK4, perturb centriole replication, result in mitosis disorder and induce cell apoptosis in breast cancer cells. Moreover 14i demonstrated significant antitumor efficacy in the MDA-MB-468 and MDA-MB-231 xenograft models. This study suggested that this concise chemotype would represent a promising scaffold of PLK4 inhibitors for cancer therapy and 14i would be an attractive lead compound for further optimization and evaluation.
Co-reporter:Zhihao Liu;Wei Wei;Lu Xiong;Qiang Feng;Yaojie Shi;Ningyu Wang;Luoting Yu
New Journal of Chemistry (1998-Present) 2017 vol. 41(Issue 8) pp:3172-3176
Publication Date(Web):2017/04/10
DOI:10.1039/C6NJ03984G
trans-Arylvinylboronate derivatives are important synthesis blocks in natural products, pharmaceuticals and organic materials. There are only a few reaction conditions that could selectively provide trans-arylvinylboronates by Heck coupling of pinacol vinylboronate and aryl halides. Here we report an efficient and versatile method of palladium catalyzed cross-coupling between pinacol vinylboronate and various aryl or hetaryl bromides to obtain the corresponding trans-(het)arylvinylboronates in excellent yields and selectivity. 30 examples have been synthesized using this protocol which offers an alternative method to prepare these useful building blocks.
Co-reporter:Li Liu;Yongxia Zhu;Zhihao Liu;Tinghong Ye;Weiqiong Zuo
Molecular Diversity 2017 Volume 21( Issue 1) pp:125-136
Publication Date(Web):2017 February
DOI:10.1007/s11030-016-9707-6
The bromodomain and extra-terminal proteins (BETs), in particular BRD4, has been reported to play important roles in cancer, inflammation, obesity, cardiovascular disease, and neurological disorders. In this paper, a series of benzomorpholinone derivatives were synthesized and biologically evaluated as BETs inhibitors. Detailed structure–activity relationship studies led to the discovery of several new potent compounds, of which 15h and 15i displayed \(\text {IC}_{50}\) values of 2.8 and 4.5 \({\upmu }\text {M}\) against BRD4 (D1), respectively, and showed good anti-proliferation activities against four hematologic malignancies cell lines at low-micromolar concentrations, including MV4-11, OCI-LY10, Pfeifer, and Su-DHL-6 cells. This chemotype could be further optimized with respect to its potency and drug-like properties in the future.
Co-reporter:Wei-Qiong Zuo, Ning-Yu Wang, Yong-xia Zhu, Li Liu, Kun-Jie Xiao, Li-Dan Zhang, Chao Gao, Zhi-Hao Liu, Xin-Yu You, Yao-Jie Shi, Cui-Ting Peng, Kai Ran, Hong Tang and Luo-Ting Yu
RSC Advances 2016 vol. 6(Issue 46) pp:40277-40286
Publication Date(Web):11 Apr 2016
DOI:10.1039/C6RA01179A
A new series of HCV inhibitors based on a 2-(thieno[2,3-b]pyridin-2-yl)-1,3,4-oxadiazole scaffold was developed. Detailed SAR investigations revealed the HCV inhibitory activity was sensitive to the size of C5, the C6-fused ring, and the size and flexibility of C5′ cycloalkane, which led to the identification of several compounds with potent inhibitory activity against HCV genotype 1b replicon. The most potent compound 10d showed ∼100-fold improvement in potency compared with compound 1, with an EC50 of 0.039 μM, but without obvious cytotoxicity in vitro.
Co-reporter:Tiantao Gao, Lidan Zhang, Yongxia Zhu, Xuejiao Song, Qiang Feng, Qian Lei, Suxia Shi, Hongxia Deng, Menghua Xiong, Xinyu You, Weiqiong Zuo, Li Liu, Cuiting Peng, Ningyu Wang, Tinghong Ye, Yong Xia and Luoting Yu
RSC Advances 2016 vol. 6(Issue 34) pp:28512-28521
Publication Date(Web):03 Mar 2016
DOI:10.1039/C6RA00618C
The histone methyltransferase enhancer of zeste homolog 2 (EZH2) has been reported to be overexpressed in a variety of cancers and is associated with tumor malignancy. This is mainly because EZH2 catalyzes the hypertrimethylation of histone 3 at lysine 27 (H3K27) at the promoter of target genes, leading to the silencing of downstream tumor suppressor genes. Hence, blocking its catalytic function may be a therapeutic strategy for the treatment of tumors which over-express or have a gain-of-function mutation in EZH2, such as lymphomas. Here, we reported a novel, selective, small-molecule inhibitor of EZH2 and EZH1 synthesized by us, ZLD1122, which inhibited both EZH1 and wild type and mutant EZH2 activities with nanomolar potency. ZLD1122 significantly inhibited intracellular H3K27 trimethylation without affecting the levels of H3, H3K9me3, and H3K4me3, indicating its selective inhibition of polycomb repressive complex 2 (PRC2) methyl catalytic function. Moreover, ZLD1122 induced G0/G1 phase arrest in diffuse large B cell lymphoma (DLBCL) cells in a dose-dependent manner via downregulation of cyclinE and CDK4 as well as upregulation of p21 and cyclinD1. Furthermore, it induced apoptosis and loss of mitochondrial membrane potential (Δψm), and elevated the levels of cleaved caspase-9 in Su-DHL-6 and Pfeiffer cells, suggesting that ZLD1122 suppresses the viability of DLBCL cells by inducing caspase-mediated intrinsic apoptosis. Taken together, these data demonstrated that ZLD1122, owing to its pharmacologically inhibitory activity against EZH2, could be a promising agent for the treatment of lymphomas with EZH2 gain-of-function mutations.
Co-reporter:Kai Ran, Chao Gao, Hongxia Deng, Qian Lei, Xinyu You, Ningyu Wang, Yaojie Shi, Zhihao Liu, Wei Wei, Cuiting Peng, Lu Xiong, Kunjie Xiao, Luoting Yu
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 15) pp:3669-3674
Publication Date(Web):1 August 2016
DOI:10.1016/j.bmcl.2016.05.088
The emergence of antibiotic resistant pathogens is an ongoing main problem in the therapy of bacterial infections. In order to develop promising antitubercular and antibacterial lead compounds, we designed and synthesized a new series of derivatives of 2-aminothiazole conjugated nitrofuran with activities against both Mycobacterium tuberculosis and Staphylococcus aureus. Eight compounds 12e, 12k, 12l, 12m, 18a, 18d, 18e, and 18j emerged as promising antitubercular agents. Structure–activity relationships (SARs) were discussed and showed that the derivatives substituted at the position-3 of benzene of 5-nitro-N-(4-phenylthiazol-2-yl)furan-2-carboxamide exhibited superior potency. The most potent compound 18e, substituted with benzamide at this position, displayed minimum inhibitory concentrations (MICs) of 0.27 μg/mL against Mtb H37Ra and 1.36 μg/mL against S. aureus. Furthermore, compound 18e had no obvious cytotoxicity to normal Vero cells (IC50 = 50.2 μM). The results suggest that the novel scaffolds of aminothiazole conjugated nitrofuran would be a promising class of potent antitubercular and antimicrobial agents.
Co-reporter:Ning-Yu Wang; Ying Xu; Wei-Qiong Zuo; Kun-Jie Xiao; Li Liu; Xiu-Xiu Zeng; Xin-Yu You; Li-Dan Zhang; Chao Gao; Zhi-Hao Liu; Ting-Hong Ye; Yong Xia; Ying Xiong; Xue-Jiao Song; Qian Lei; Cui-Ting Peng; Hong Tang; Sheng-Yong Yang; Yu-Quan Wei
Journal of Medicinal Chemistry 2015 Volume 58(Issue 6) pp:2764-2778
Publication Date(Web):February 24, 2015
DOI:10.1021/jm501934n
The design, synthesis, and SAR studies of novel inhibitors of HCV NS4B based on the imidazo[2,1-b]thiazole scaffold were described. Optimization of potency with respect to genotype 1b resulted in the discovery of two potent leads 26f (EC50 = 16 nM) and 28g (EC50 = 31 nM). The resistance profile studies revealed that 26f and 28g targeted HCV NS4B, more precisely the second amphipathic α helix of NS4B (4BAH2). Cross-resistance between our 4BAH2 inhibitors and other direct-acting antiviral agents targeting NS3/4A, NS5A, and NS5B was not observed. For the first time, the synergism of a series of combinations based on 4BAH2 inhibitors was evaluated. The results demonstrated that our 4BAH2 inhibitor 26f was synergistic with NS3/4A inhibitor simeprevir, NS5A inhibitor daclatasvir, and NS5B inhibitor sofosbuvir, and it could also reduce the dose of these drugs at almost all effect levels. Our study suggested that favorable effects could be achieved by combining 4BAH2 inhibitors such as 26f with these approved drugs and that new all-oral antiviral combinations based on 4BAH2 inhibitors were worth developing to supplement or even replace current treatment regimens for curing HCV infection.
Co-reporter:Qian Lei, Lidan Zhang, Yong Xia, Tinghong Ye, Fangfang Yang, Yongxia Zhu, Xuejiao Song, Ningyu Wang, Ying Xu, Xiaowei Liu and Luoting Yu
RSC Advances 2015 vol. 5(Issue 52) pp:41341-41351
Publication Date(Web):28 Apr 2015
DOI:10.1039/C5RA05387K
Hepatocellular carcinoma is the fifth most common cancer and durable responses in conventional treatments are limited so researchers have been devoted to developing new anti-HCC agents. Benzothiazole derivatives are known for various biological activities and have received considerable attention in cancer therapy, hence we designed and synthesized a novel potent benzothiazole compound 2-chloro-N-(2-(2-(2-morpholino-2-oxoethyl)thio)-2,3-dihydrobenzo[d]thiazol-6-yl)acetamide (SKLB826) and further investigated the biological activities against cancer. The results suggested that SKLB826 showed growth inhibition against a broad spectrum of human cancer cells, especially human HCC cell lines, in a dose-dependent manner and induced G2/M phase arrest via down-regulating the CDK1, cyclinA2 and cdc25c protein levels. SKLB826 could also induce apoptosis of HCC cells via decreasing the expression of Bcl-2 and increasing the levels of BAX and cleaved caspase-3, 9. Moreover, after treatment with SKLB826, the change of ROS level and ΔΨm suggested that SKLB826 might induce apoptosis through an intrinsic mitochondrial apoptotic pathway. Furthermore, SKLB826 could suppress tumor growth in the HepG2 xenograft model without inducing any notable major organ-related toxicity, suggesting that SKLB826 may be a potential candidate for HCC therapy.
Co-reporter:Cui-Ting Peng, Chao Gao, Ning-Yu Wang, Xin-Yu You, Li-Dan Zhang, Yong-Xia Zhu, Ying Xv, Wei-Qiong Zuo, Kai Ran, Hong-Xia Deng, Qian Lei, Kun-Jie Xiao, Luo-Ting Yu
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 7) pp:1373-1376
Publication Date(Web):1 April 2015
DOI:10.1016/j.bmcl.2015.02.061
Tuberculosis (TB) remains a major human health problem. New therapeutic antitubercular agents are urgent needed to control the global tuberculosis pandemic. We synthesized a new series of 4-carbonyl piperazine substituted 1,3-benzothiazin-4-one derivatives and evaluated their anti-mycobacterial activities against Mycobacterium tuberculosis H37Ra as well as their druggabilities. The results showed that most of these derivatives, especially the compounds with simple alkyl side chains, exhibited good antitubercular activities and favorable aqueous solubilities with no obvious cytotoxicity. It suggested that the 4-carbonyl piperazine substituents in benzothiazinone scaffold were well tolerated, in which the compound 8h, with an antitubercular activity of MIC 0.008 μM, exhibited an excellent aqueous solubility of 104 μg/mL, which was 100-fold better than the potent DprE1 inhibitor Comp.1 (BTZ038), also more soluble than PBTZ169.
Co-reporter:Yong Luo, Yongxia Zhu, Kai Ran, Zhihao Liu, Ningyu Wang, Qiang Feng, Jun Zeng, Lidan Zhang, Bing He, Tinghong Ye, Shirui Zhu, Xiaolong Qiu and Luoting Yu
MedChemComm 2015 vol. 6(Issue 6) pp:1036-1042
Publication Date(Web):13 Apr 2015
DOI:10.1039/C4MD00573B
In this study, a series of novel N-(4-phenylthiazol-2-yl)cinnamamide derivatives (7a–8n) were synthesized and evaluated for their anti-proliferative activities in vitro by MTT assay and a possible antitumor mechanism was also explored. SAR analysis showed that steric effects played an important role on the anti-tumor activity. The most potent analogue 8f showed excellent inhibitions on the K562, Bel7402, A549 and Jurkat cells ranging from sub-micromolar to nanomolar concentration. Compound 8f inhibited Jurkat cells with an IC50 value of 0.035 μM with no apparent toxicity in different non-cancerous cells. Furthermore, it was suggested that the possible mechanism of 8f might be associated with inducing cancer cell apoptosis following flow cytometer analysis and Hoechst 33358 staining assays.
Co-reporter:Yong Luo, Yongxia Zhu, Kai Ran, Zhihao Liu, Ningyu Wang, Qiang Feng, Jun Zeng, Lidan Zhang, Bing He, Tinghong Ye, Shirui Zhu, Xiaolong Qiu and Luoting Yu
MedChemComm 2015 vol. 6(Issue 7) pp:1404-1404
Publication Date(Web):15 Jun 2015
DOI:10.1039/C5MD90027A
Correction for 'Synthesis and biological evaluation of N-(4-phenylthiazol-2-yl)cinnamamide derivatives as novel potential anti-tumor agents' by Yong Luo et al., Med. Chem. Commun., 2015, 6, 1036.
Co-reporter:Ying Xu, Ning-Yu Wang, Xue-Jiao Song, Qian Lei, Ting-Hong Ye, Xin-Yu You, Wei-Qiong Zuo, Yong Xia, Li-Dan Zhang, Luo-Ting Yu
Bioorganic & Medicinal Chemistry 2015 23(15) pp: 4333-4343
Publication Date(Web):
DOI:10.1016/j.bmc.2015.06.033
Co-reporter:X Peng, G Xie, Z Wang, H Lin, T Zhou, P Xiang, Y Jiang, S Yang, Y Wei, L Yu and Y Zhao
Cell Death & Disease 2014 5(3) pp:e1143
Publication Date(Web):2014-03-01
DOI:10.1038/cddis.2014.107
Small-molecule inhibitors are an attractive therapeutic approach for most types of human cancers. SKLB-163, a novel benzothiazole-2-thiol derivative, was developed via computer-aided drug design and de novo synthesis. MTT assay showed it had potent anti-proliferative activity on various human cancer cells. Treatment of cancer cells with SKLB-163 induced obvious apoptosis and inhibited proliferation in vitro. SKLB-163 administered p.o. showed a marked antitumor activity in vivo. Proteomic techniques were employed to identify possible drug target proteins. The data showed molecular mechanism of action might be involved in downregulation of RhoGDI, which finally contributed to increased apoptosis and inhibited proliferation. These findings provided the potential value of SKLB-163 as a novel candidate antitumor drug.
Co-reporter:Ning-Yu Wang, Wei-Qiong Zuo, Ying Xu, Chao Gao, Xiu-Xiu Zeng, Li-Dan Zhang, Xin-Yu You, Cui-Ting Peng, Yang Shen, Sheng-Yong Yang, Yu-Quan Wei, Luo-Ting Yu
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 6) pp:1581-1588
Publication Date(Web):15 March 2014
DOI:10.1016/j.bmcl.2014.01.075
Current treatment for hepatitis C is barely satisfactory, there is an urgent need to develop novel agents for combating hepatitis C virus infection. This study discovered a new class of thieno[2,3-b]pyridine derivatives as HCV inhibitors. First, a hit compound characterized by a thienopyridine core was identified in a cell-based screening of our privileged small molecule library. And then, structure activity relationship study of the hit compound led to the discovery of several potent compounds without obvious cytotoxicity in vitro (12c, EC50 = 3.3 μM, SI >30.3, 12b, EC50 = 3.5 μM, SI >28.6, 10l, EC50 = 3.9 μM, SI >25.6, 12o, EC50 = 4.5 μM, SI >22.2, respectively). Although the mechanism of them had not been clearly elucidated, our preliminary optimization of this class of compounds had provided us a start point to develop new anti-HCV agents.SAR investigation on five regions of thieno[2,3-b]pyridine derivatives led to the discovery of several promising compounds which exhibited low micromolar potency against HCV replicon.
Co-reporter:Tinghong Ye;Deliang Li;Yuquan Wei;Guobo Shen;Luoting Yu;Yongmei Xie;Ying Xiong;Ningyu Wang;Lifeng Zhao;Can Luo;Jun Zeng;Zhiyao He;Xiang Gao;Min Luo;Bin Shao;Xuejiao Song;Yong Xia;Sisi He;Tao Yin;Xiawei Wei
Breast Cancer Research and Treatment 2014 Volume 143( Issue 3) pp:435-446
Publication Date(Web):2014/02/01
DOI:10.1007/s10549-013-2829-y
Aberrant fibroblast growth factor (FGF) and FGF receptor (FGFR) system have been associated with breast cancer. The objectives of our study were to investigate the effects and mechanisms of FGFR inhibition on tumor growth and metastasis on breast cancer. Our studies showed that the FGFR inhibitor PD173074 decreased the viability of several human breast cancer cells, as well as 4T1 murine mammary tumor cells. Therefore, we chose 4T1 cells to study PD173074’s antitumor mechanism. Flow cytometry showed that PD173074 induced 4T1 cell apoptosis in a concentration-dependent manner. Western blot demonstrated that PD173074-induced apoptosis was correlated with the inhibition of Mcl-1 and survivin. Moreover, PD173074 also significantly increased the ratio of Bax/Bcl-2. PD173074 could also block 4T1 cell migration and invasion in vitro. In 4T1 tumor-bearing mice, PD173074 significantly inhibited tumor growth without obvious side effects. Meanwhile, PD173074 functionally reduced microvessel density and proliferation index and induced tumor apoptosis. Importantly, we found that FGFR inhibition by PD173074 reduced myeloid-derived suppressor cells (MDSCs) in the blood, spleens and tumors, accompanied by the increased infiltration of CD4+ and CD8+ T cells in the spleens and tumors. Furthermore, PD173074 significantly inhibited breast tumor metastasis to the lung of inoculated 4T1 breast cancer cells, which was accompanied by a reduction in MDSCs. Our findings suggested that FGFR inhibition could delay breast tumor progression, impair lung metastasis and break immunosuppression by effecting on tumor microenvironment, which may provide a promising therapeutic approach for breast cancer patient.
Co-reporter:Lifeng Zhao, Xiao Li, Lidan Zhang, Tinghong Ye, Yongxia Zhu, Yuquan Wei, Shengyong Yang, Luoting Yu
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 8) pp:2293-2297
Publication Date(Web):15 April 2013
DOI:10.1016/j.bmcl.2013.02.076
Inducing apoptosis is a promising therapeutic approach to overcome cancer. In this study, 30 compounds were synthesized and evaluated for their antiproliferative activity against three tumor cell lines in vitro: A875, H460 and Hela cancer cells by the MTT assay. The most potent analogue 7a, a novel compound was first reported by our group, inhibited the proliferation of A875 cells with an IC50 value of 98 nM. Flow cytometry analysis and morphological analysis suggested that compound 7a had potential anticancer efficacy via G2/M cell cycle arrest, which could be attributed to its proliferation and apoptosis, and also in a concentration-dependent manner. The SAR analysis indicated that the substituents R2 played a crucial role in the antiproliferation activity.
Co-reporter:Chao Gao, Ting-Hong Ye, Ning-Yu Wang, Xiu-Xiu Zeng, Li-Dan Zhang, Ying Xiong, Xin-Yu You, Yong Xia, Ying Xu, Cui-Ting Peng, Wei-Qiong Zuo, Yuquan Wei, Luo-Ting Yu
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 17) pp:4919-4922
Publication Date(Web):1 September 2013
DOI:10.1016/j.bmcl.2013.06.069
N-Alkyl and heterocycle substituted 1,3-benzothiazin-4-one (BTZ) derivatives were synthesized. The anti-mycobacterial activities of these compounds were evaluated by determination of minimal inhibitory concentration (MIC) for Mycobacterium tuberculosis H37Ra and M. tuberculosis H37Rv. It was found that an extended or branched alkyl chain analog could enhance the potency, and activities of N-alkyl substituted BTZs were not affected by either nitro or trifluoromethyl at 6-position. Trifluoromethyl plays an important role in maintaining anti-tubercular activity in the piperazine or piperidine analogs. Compound 8o, which contains an azaspirodithiolane group, showed a MIC of 0.0001 μM against M. tuberculosis H37Rv, 20-fold more potent than BTZ043 racemate. These results suggested that the volume and lipophilicity of the substituents were important in maintaining activity. In addition, compound 8o was nontoxic to Vero cells and orally bioavailable in a preliminary pharmacokinetics study.The synthesis and SAR profile of N-alkyl and heterocycle substituted 1,3-benzothiazin-4-one (BTZ) derivatives are described. The most potent compound 8o exhibits a MIC of 0.0001 μM against Mycobacterium tuberculosis H37Rv, 20-fold more potent than BTZ043 racemate.
Co-reporter:Zhao Wang, Xuan-Hong Shi, Jia Wang, Tian Zhou, You-Zhi Xu, Ting-Ting Huang, Yan-Fang Li, Ying-Lan Zhao, Li Yang, Sheng-Yong Yang, Luo-Ting Yu, Yu-Quan Wei
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 4) pp:1097-1101
Publication Date(Web):15 February 2011
DOI:10.1016/j.bmcl.2010.12.124
A series of novel benzothiazole-2-thiol derivatives were synthesized, and their anti-proliferative activities on HepG2 and MCF-7 cells were investigated. Most compounds had inhibitory effects on cell growth, and some of them were more effective than cisplatin. Compounds 6m and 6t displayed good inhibitory activities against a panel of different types of human cancer cell lines, with IC50 values in the low micromolar range. Further biological evaluation indicated that 6m induced apoptosis in HepG2 cancer cells. Structure–activity relationships were also proposed.A series of novel benzothiazole-2-thiol derivatives were synthesized and evaluated for antitumor. The most potent compound 6m exhibited good inhibitory activities against a panel of different types of human cancer cell lines with IC50 values in the low micromolar range and induced apoptosis in HepG2 cancer cells.

![PROPAN-2-YL (2S)-2-[[[(2R,3R,4R,5R)-5-(2,4-DIOXOPYRIMIDIN-1-YL)-4-FLUORO-3-HYDROXY-4-METHYLOXOLAN-2-YL]METHOXY-PHENOXYPHOSPHORYL]AMINO]PROPANOATE](http://img.cochemist.com/ccimg/1190400/1190307-88-0.png)
![PROPAN-2-YL (2S)-2-[[[(2R,3R,4R,5R)-5-(2,4-DIOXOPYRIMIDIN-1-YL)-4-FLUORO-3-HYDROXY-4-METHYLOXOLAN-2-YL]METHOXY-PHENOXYPHOSPHORYL]AMINO]PROPANOATE](http://img.cochemist.com/ccimg/1190400/1190307-88-0_b.png)


![7-bromo-2-(4-nitrophenyl)imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/947600/947533-60-0.png)
![7-bromo-2-(4-nitrophenyl)imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/947600/947533-60-0_b.png)
![2-(4-nitrophenyl)-8-(trifluoromethyl)imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/944600/944581-02-6.png)
![2-(4-nitrophenyl)-8-(trifluoromethyl)imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/944600/944581-02-6_b.png)
![Benzenamine, 4-(6-fluoroimidazo[1,2-a]pyridin-2-yl)-](http://img.cochemist.com/ccimg/774300/774239-27-9.png)
![Benzenamine, 4-(6-fluoroimidazo[1,2-a]pyridin-2-yl)-](http://img.cochemist.com/ccimg/774300/774239-27-9_b.png)
![Imidazo[1,2-a]pyridine, 6-fluoro-2-(4-nitrophenyl)-](http://img.cochemist.com/ccimg/774300/774239-26-8.png)
![Imidazo[1,2-a]pyridine, 6-fluoro-2-(4-nitrophenyl)-](http://img.cochemist.com/ccimg/774300/774239-26-8_b.png)
![Benzenamine, 4-(6-chloroimidazo[1,2-a]pyridin-2-yl)-](http://img.cochemist.com/ccimg/774300/774239-24-6.png)
![Benzenamine, 4-(6-chloroimidazo[1,2-a]pyridin-2-yl)-](http://img.cochemist.com/ccimg/774300/774239-24-6_b.png)
![4-(6-BROMOIMIDAZO[1,2-A]PYRIDIN-2-YL)ANILINE](http://img.cochemist.com/ccimg/774300/774239-23-5.png)
![4-(6-BROMOIMIDAZO[1,2-A]PYRIDIN-2-YL)ANILINE](http://img.cochemist.com/ccimg/774300/774239-23-5_b.png)
![Benzenamine, 4-(6-iodoimidazo[1,2-a]pyridin-2-yl)-](http://img.cochemist.com/ccimg/774300/774238-56-1.png)
![Benzenamine, 4-(6-iodoimidazo[1,2-a]pyridin-2-yl)-](http://img.cochemist.com/ccimg/774300/774238-56-1_b.png)
![3-METHYL-N-{[2-(PENTAFLUOROPHENYL)-1,3-BENZOXAZOL-5-YL]CARBAMOTHIOYL}BUTANAMIDE](/data/chemimg/631900/625370-80-1.png)
![3-METHYL-N-{[2-(PENTAFLUOROPHENYL)-1,3-BENZOXAZOL-5-YL]CARBAMOTHIOYL}BUTANAMIDE](/data/chemimg/631900/625370-80-1_b.png)
![<br>(3-Amino-6-(thiophen-2-yl)-4-(trifluoromethyl)thieno[2,3-b]pyridin-2-yl)(mo rpholino)methanone](http://img.cochemist.com/ccimg/400900/400863-57-2.png)
![<br>(3-Amino-6-(thiophen-2-yl)-4-(trifluoromethyl)thieno[2,3-b]pyridin-2-yl)(mo rpholino)methanone](http://img.cochemist.com/ccimg/400900/400863-57-2_b.png)
![<br>3-Amino-N,N-diethyl-6-phenyl-4-(trifluoromethyl)thieno[2,3-b]pyridine-2-car boxamide](http://img.cochemist.com/ccimg/400900/400863-50-5.png)
![<br>3-Amino-N,N-diethyl-6-phenyl-4-(trifluoromethyl)thieno[2,3-b]pyridine-2-car boxamide](http://img.cochemist.com/ccimg/400900/400863-50-5_b.png)
![4-(8-METHYLIMIDAZO[1,2-A]PYRIDIN-2-YL)ANILINE](http://img.cochemist.com/ccimg/365600/365565-88-4.png)
![4-(8-METHYLIMIDAZO[1,2-A]PYRIDIN-2-YL)ANILINE](http://img.cochemist.com/ccimg/365600/365565-88-4_b.png)
![Imidazo[1,2-a]pyridine,6-bromo-2-(4-nitrophenyl)-](http://img.cochemist.com/ccimg/322000/321945-25-9.png)
![Imidazo[1,2-a]pyridine,6-bromo-2-(4-nitrophenyl)-](http://img.cochemist.com/ccimg/322000/321945-25-9_b.png)


![Ethyl 3-amino-4-(trifluoromethyl)-6-phenylfuro[2,3-b]pyridine-2-carboxylate](http://img.cochemist.com/ccimg/160500/160436-48-6.png)
![Ethyl 3-amino-4-(trifluoromethyl)-6-phenylfuro[2,3-b]pyridine-2-carboxylate](http://img.cochemist.com/ccimg/160500/160436-48-6_b.png)


![4-(Imidazo[1,2-a]pyridin-2-yl)aniline](http://img.cochemist.com/ccimg/139800/139705-74-1.png)
![4-(Imidazo[1,2-a]pyridin-2-yl)aniline](http://img.cochemist.com/ccimg/139800/139705-74-1_b.png)
![Imidazo[2,1-b]thiazole-6-carboxylic acid, 2-methyl-, ethyl ester](http://img.cochemist.com/ccimg/120200/120107-64-4.png)
![Imidazo[2,1-b]thiazole-6-carboxylic acid, 2-methyl-, ethyl ester](http://img.cochemist.com/ccimg/120200/120107-64-4_b.png)
![Imidazo[1,2-a]pyridine, 6-iodo-2-(4-nitrophenyl)-](http://img.cochemist.com/ccimg/118100/118000-67-2.png)
![Imidazo[1,2-a]pyridine, 6-iodo-2-(4-nitrophenyl)-](http://img.cochemist.com/ccimg/118100/118000-67-2_b.png)
![Imidazo[1,2-a]pyridine, 6-chloro-2-(4-nitrophenyl)-](http://img.cochemist.com/ccimg/118100/118000-62-7.png)
![Imidazo[1,2-a]pyridine, 6-chloro-2-(4-nitrophenyl)-](http://img.cochemist.com/ccimg/118100/118000-62-7_b.png)


![3-Amino-6-phenyl-4-(trifluoromethyl)thieno[2,3-b]pyridine-2-carboxylic acid](http://img.cochemist.com/ccimg/105000/104960-56-7.png)
![3-Amino-6-phenyl-4-(trifluoromethyl)thieno[2,3-b]pyridine-2-carboxylic acid](http://img.cochemist.com/ccimg/105000/104960-56-7_b.png)
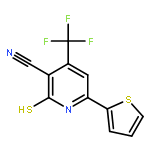
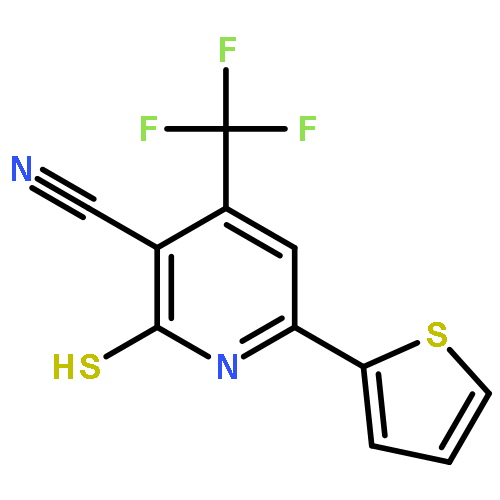
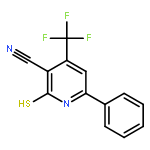
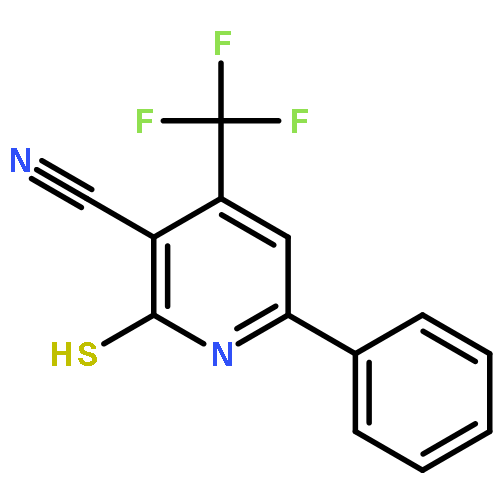
![4-(7-Methylimidazo[1,2-a]pyridin-2-yl)aniline](http://img.cochemist.com/ccimg/64800/64730-34-3.png)
![4-(7-Methylimidazo[1,2-a]pyridin-2-yl)aniline](http://img.cochemist.com/ccimg/64800/64730-34-3_b.png)


![Benzenamine, 4-imidazo[1,2-a]pyrimidin-2-yl-](http://img.cochemist.com/ccimg/58700/58609-93-1.png)
![Benzenamine, 4-imidazo[1,2-a]pyrimidin-2-yl-](http://img.cochemist.com/ccimg/58700/58609-93-1_b.png)
![THIENO[2,3-B]PYRIDINE-2-CARBOXYLIC ACID, 3-AMINO-4-METHYL-6-PHENYL-](http://img.cochemist.com/ccimg/58400/58327-78-9.png)
![THIENO[2,3-B]PYRIDINE-2-CARBOXYLIC ACID, 3-AMINO-4-METHYL-6-PHENYL-](http://img.cochemist.com/ccimg/58400/58327-78-9_b.png)
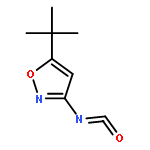
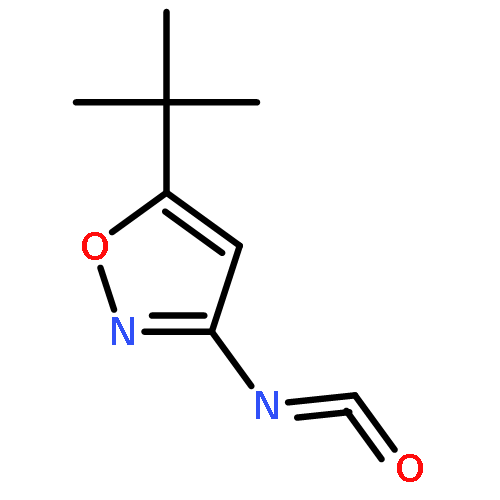
![Benzenamine, 4-benzo[b]thien-2-yl-](http://img.cochemist.com/ccimg/54500/54492-95-4.png)
![Benzenamine, 4-benzo[b]thien-2-yl-](http://img.cochemist.com/ccimg/54500/54492-95-4_b.png)
![Benzo[b]thiophene, 2-(4-nitrophenyl)-](http://img.cochemist.com/ccimg/54500/54492-94-3.png)
![Benzo[b]thiophene, 2-(4-nitrophenyl)-](http://img.cochemist.com/ccimg/54500/54492-94-3_b.png)
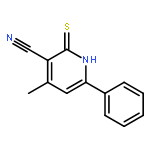
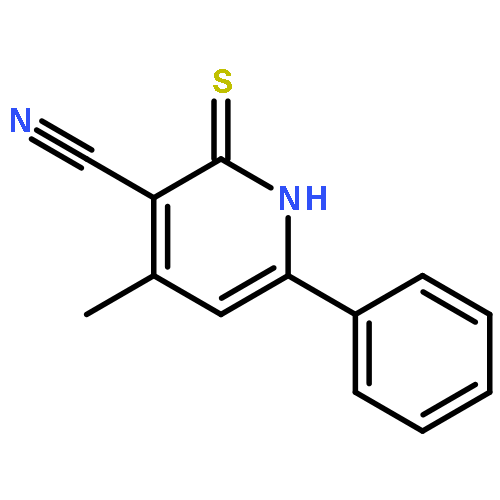


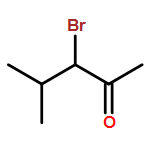
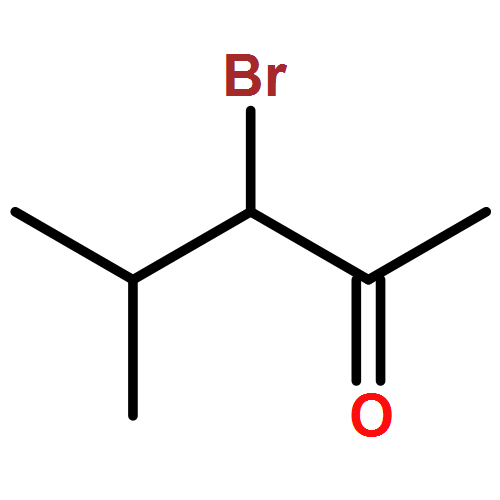
![2-(4-Nitrophenyl)imidazo[1,2-a]pyrimidine](http://img.cochemist.com/ccimg/28300/28266-96-8.png)
![2-(4-Nitrophenyl)imidazo[1,2-a]pyrimidine](http://img.cochemist.com/ccimg/28300/28266-96-8_b.png)
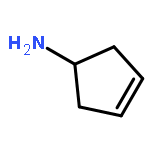
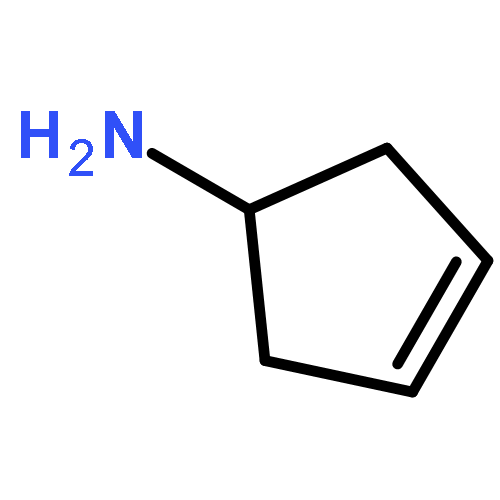


![4-(Benzo[d]oxazol-2-yl)aniline](http://img.cochemist.com/ccimg/21000/20934-81-0.png)
![4-(Benzo[d]oxazol-2-yl)aniline](http://img.cochemist.com/ccimg/21000/20934-81-0_b.png)
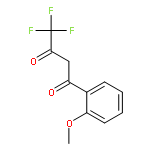
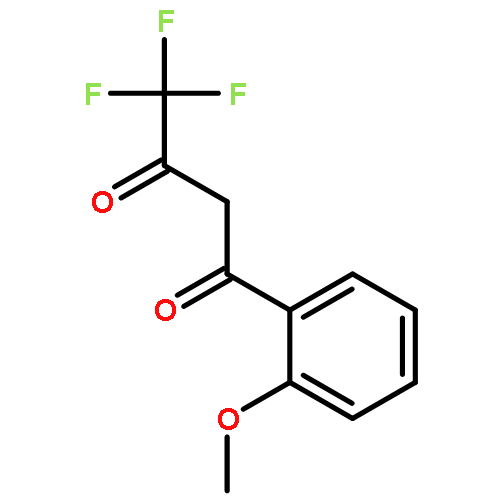
![N-[(4-Nitrophenyl)sulfonyl]glycine](http://img.cochemist.com/ccimg/15100/15054-44-1.png)
![N-[(4-Nitrophenyl)sulfonyl]glycine](http://img.cochemist.com/ccimg/15100/15054-44-1_b.png)
![5,6,7,8-Tetrahydro-4H-cyclohepta[d]thiazol-2-amine](http://img.cochemist.com/ccimg/14300/14292-44-5.png)
![5,6,7,8-Tetrahydro-4H-cyclohepta[d]thiazol-2-amine](http://img.cochemist.com/ccimg/14300/14292-44-5_b.png)
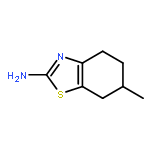
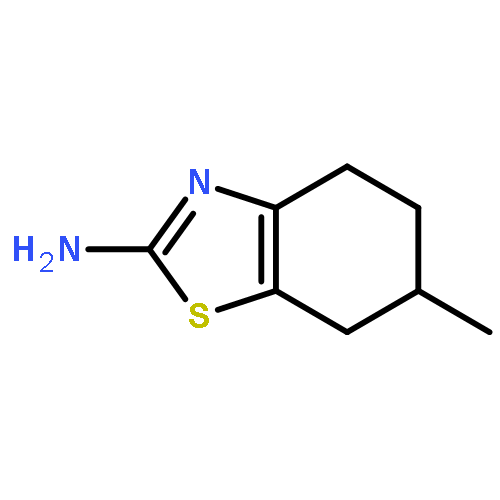
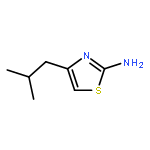
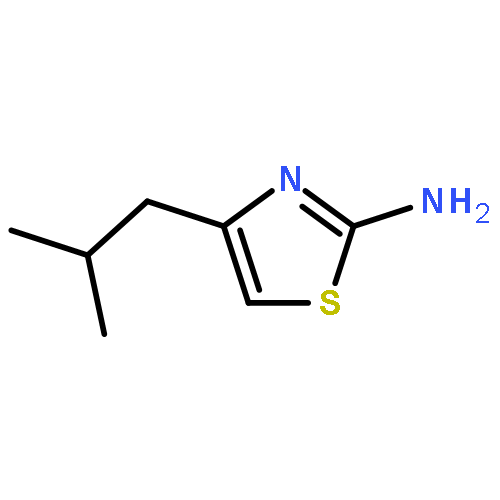
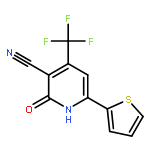
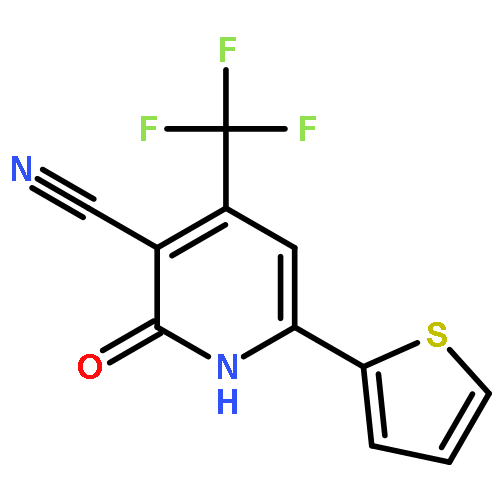
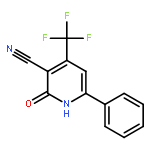
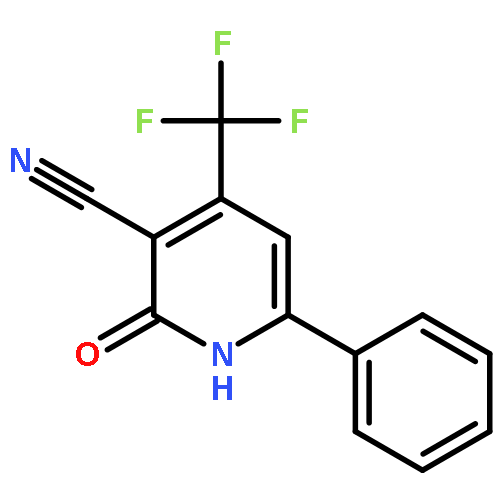


![Imidazo[1,2-a]pyridine, 7-methyl-2-(4-nitrophenyl)-](http://img.cochemist.com/ccimg/1000/961-82-0.png)
![Imidazo[1,2-a]pyridine, 7-methyl-2-(4-nitrophenyl)-](http://img.cochemist.com/ccimg/1000/961-82-0_b.png)
![IMIDAZO[1,2-A]PYRIDINE, 8-METHYL-2-(4-NITROPHENYL)-](http://img.cochemist.com/ccimg/900/893-16-3.png)
![IMIDAZO[1,2-A]PYRIDINE, 8-METHYL-2-(4-NITROPHENYL)-](http://img.cochemist.com/ccimg/900/893-16-3_b.png)
![Imidazo[1,2-a]pyridine, 6-methyl-2-(4-nitrophenyl)-](http://img.cochemist.com/ccimg/900/842-73-9.png)
![Imidazo[1,2-a]pyridine, 6-methyl-2-(4-nitrophenyl)-](http://img.cochemist.com/ccimg/900/842-73-9_b.png)

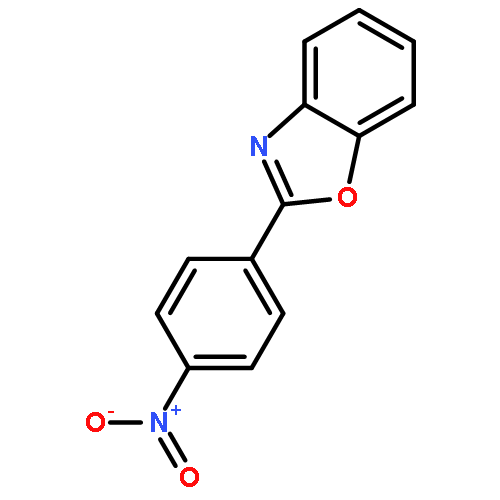






![1H-Benzimidazole,1-[(4-chlorophenyl)methyl]-2-(1-pyrrolidinylmethyl)-](http://img.cochemist.com/ccimg/500/442-52-4.png)
![1H-Benzimidazole,1-[(4-chlorophenyl)methyl]-2-(1-pyrrolidinylmethyl)-](http://img.cochemist.com/ccimg/500/442-52-4_b.png)

