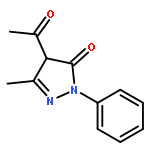
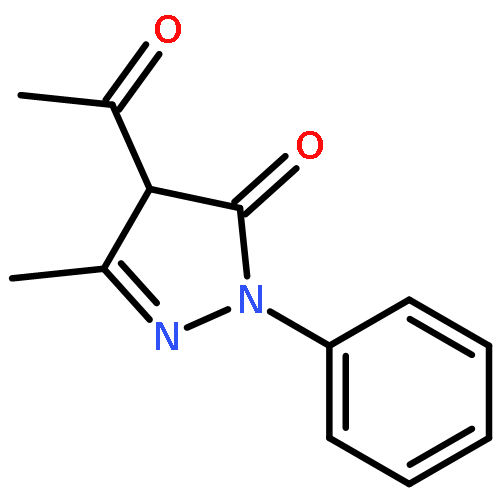
Co-reporter:Yunhe Li, Alexander M. Kirillov, Ran Fang, and Lizi Yang
Organometallics March 27, 2017 Volume 36(Issue 6) pp:1164-1164
Publication Date(Web):March 15, 2017
DOI:10.1021/acs.organomet.7b00042
This study reports a detailed theoretical analysis of the mechanisms and chemoselectivity for the formation of benzo[b]fluorenes or benzofulvenes from propargyl esters catalyzed by an organometallic Au(I) complex. Three different substitution patterns within the 1,5-diyne ester substrates were explored to rationalize the reaction mechanism and chemoselectivity. DFT calculations reveal that the title reaction proceeds through four main steps: (i) 1,3-acyl-shift, (ii) 6-endo-dig or 5-exo-dig cyclization, (iii) Friedel–Crafts-type, and (iv) proton transfer, with step (ii) being rate-determining in all studied pathways. In the absence of substituents at the aromatic rings of the substrate (R = H), the 6-endo-dig cyclization is favored. In turn, in the presence of one strong electron-donating substituent at the backbone (R = OCH3) of the substrate, the 5-exo-dig cyclization is favored. Besides, a modification of the substrate’s acetyl group by a pivaloyl group leads to an activation barrier difference between the 6-endo-dig and 5-exo-dig cyclizations, which increases and suppresses the formation of benzofulvenes. The obtained theoretical data are in a very good agreement with prior experimental evidence, suggesting that the substituent plays a crucial role in the outcome of the final product. High chemoselectivity can be explained by the hindrance (torsional strain) along the forming C–C bond and the carbocation stability provided by substituents.
Co-reporter:Xiaoxiao Wei, Ran Fang and Lizi Yang
Catalysis Science & Technology 2015 vol. 5(Issue 6) pp:3352-3362
Publication Date(Web):20 Apr 2015
DOI:10.1039/C5CY00407A
The reaction mechanisms of N-heterocylic carbene (NHC)-catalyzed annulation to 3-pyrancarbaldehydes or cyclopentene were theoretically analyzed. Different from what has been reported for normal aldehydes or enals, our calculation results revealed that the intermediate results from the conjugate addition to the central carbon atom of the allene moiety, instead of forming the Breslow intermediate. The next step, 1,4-addition of the chalcone, resulted in the formation of an enolate anion intermediate. Then, intramolecular conjugate addition and elimination of the NHC catalyst afforded the final product. In addition, the computed results showed good agreement with the experimental evidence, suggesting that both the existence of an allene structure and charge distribution play a crucial role in the outcome of chemoselectivity and involvement of operative intermediates in the reaction pathway.
Co-reporter:Ran Fang, Xiaoxiao Wei and Lizi Yang
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 42) pp:8433-8441
Publication Date(Web):01 Sep 2014
DOI:10.1039/C4OB00894D
We report here the theoretical analysis of the mechanism and regioselectivity of gold(I) or platinum(II) catalyzed intramolecular hydroarylation to pyrrolopyridinones and pyrroloazepinones. AuPH3+ and PtCl2 have been considered to account for some experimental observations. Our calculation results indicate that in the case of cationic gold the nucleophilic attack of the pyrrole on the activated alkyne occurs in an exo-dig fashion generating a six-membered intermediate, which upon deprotonation and protodeauration forms pyrrolopyridinone. When platinum is used, an endo-dig fashion is observed generating a seven-membered intermediate. After deprotonation and protodeplatination pyrroloazepinone is formed. Whether for exo-dig (gold(I)) or endo-dig (platinum(II)) cyclization, a [1,2]-migration would not be needed.
Co-reporter:Ran Fang, Lizi Yang, and Qiang Wang
Organometallics 2014 Volume 33(Issue 1) pp:53-60
Publication Date(Web):December 18, 2013
DOI:10.1021/om400647e
We report here the theoretical analysis of the mechanism and regioselectivity of the direct insertion of silane moieties into the C–N bond of N-heterocyclic carbenes leading to eventual ring expansion and formation of diazasilinanes. Symmetrically and unsymmetrically substituted NHC have been considered to account for some experimental observations. These reaction steps include (1) Si–H bond activation of the silane at the NHC; (2) amide transfer to the silicon atom to yield a six-membered intermediate; (3) hydride or phenyl group transfer to the carbon atom to give the product. In addition, the computed results over a variety of silanes agree with experimental evidence and suggest that both steric and electronic effects play a crucial role in the regioselectivity outcome and the involvement of operative intermediates in the reaction pathway.
Co-reporter:Lizi Yang, Ran Fang
Journal of Molecular Catalysis A: Chemical 2013 Volume 379() pp:197-206
Publication Date(Web):15 November 2013
DOI:10.1016/j.molcata.2013.08.010
•Mechanisms of gold (I)-catalyzed cycloisomerization were investigated.•Three possible mechanisms for title reaction are considered.•Theoretical investigation was performed to explain the difference of reactivity.•Water-assisted [1,2]-H shift for title reaction was located.•Calculated results are consistent with the experimental observations.The mechanisms of the gold (I)-catalyzed cycloisomerization of propargylic esters leading to unsymmetrically substituted naphthalenes have been investigated using density functional theory calculations done at the B3LYP/6-31G (d, p) (SDD for Au) level of theory. Solvent effects on these reactions have been explored by calculations that included a polarizable continuum model (PCM) for the solvent (CH2Cl2). Three possible pathways which lead to forming 1,3-diene, allene or carbenoid intermediate via an unprecedented tandem sequence of [1,3]- and [1,2]-migration of two different migrating groups were proposed. Calculations suggest that the [1,3]-rearrangement is a two-step process with activation free energies below 11.0 kcal/mol for both steps. The following of [1,2]-migration reaction is also easy with an activation free energy of 20.4 kcal/mol in CH2Cl2. The next step in the catalytic cycle is a [1,2]-hydride shift, and this step is the rate-limiting step (with a calculated activation free energy of 23.2 kcal/mol) without water. In the presence of water, the direct [1,2]-hydride shift has been changed into a deprotonation/protonation process with an activation free energy of 9.7 kcal/mol. The higher activation free energies for the intramolecular 6π electrocyclic ring closure process indicate that this step became the rate-determining one. Calculations show that a water-catalyzed [1,2]-hydrogen shift adopts a proton-transport catalysis strategy, in which the acetoxy group in the substrate is critical because it acts as either a proton acceptor when one water molecule or a water cluster is involved in catalysis. Our calculated results are consistent with the experimental observations of Gevorgyan et al. for the gold (I)-catalyzed cycloisomerization of propargylic esters leading to unsymmetrically substituted naphthalenes.
Co-reporter:Ran Fang and Lizi Yang
Organometallics 2012 Volume 31(Issue 8) pp:3043-3055
Publication Date(Web):April 5, 2012
DOI:10.1021/om201159t
The mechanisms of the gold(I)-catalyzed rearrangement of homopropargyl sulfoxides have been investigated using density functional theory calculations done at the B3LYP/6-31G(d, p) (SDD for Au) level of theory. Solvent effects on these reactions have been explored by calculations that included a polarizable continuum model (PCM) for the solvent (CH2Cl2). Two plausible pathways which lead to the formation of benzothiepinones or benzothiopines via an α-carbonyl Au carbenoid through 5-exo-dig cyclization or 6-endo-dig cyclization were proposed. Our calculation results suggested the following. (1) The first step of the cycle is nucleophilic addition of the sulfoxide oxygen onto the triple bond to form an alkenyl gold intermediate through 5-exo-dig cyclization or 6-endo-dig cyclization. The alkenyl gold species is then capable of pushing out the sulfide moiety, forming an α-carbonyl Au carbenoid. Finally, α-carbonyl Au carbenoids undergo intramolecular Friedel–Crafts alkylation to produce the observed products and liberate the cationic gold(I) catalyst. (2) When the alkyne is substituted with an electron-withdrawing group, 5-exo-dig cyclization of the nucleophile onto the internal carbon of the alkyne is favored. On the other hand, when the alkyne is substituted with an alkyl group, 6-endo-dig cyclization of the nucleophile onto the internal carbon of the alkyne is favored. (3) For 6-endo-dig cyclization, an intramolecular reaction of the α-carbonyl Au carbenoid with the benzene ring was the rate-determining step. However, migration of the hydrogen atom resulting in the formation of the final product was the rate-determining step for 5-exo-dig cyclization. (4) In the presence of water, the direct [1,5]-hydride shift has been changed into a deprotonation/protonation process. A very easy deprotonation process was found in the case of water. The higher activation free energies for the protonation process indicate that this step became the rate-determining one.
Co-reporter:Ran Fang, Lizi Yang, and Qiang Wang
Organometallics 2012 Volume 31(Issue 10) pp:4020-4030
Publication Date(Web):May 11, 2012
DOI:10.1021/om300240n
The mechanisms of PtCl2-catalyzed rearrangement of cyclopropenes to allenes have been investigated using density functional theory calculations carried out at the B3LYP/6-31G(d,p) (LANL2TZ(f) for Pt) level of theory. The solvent effect was taken into account by B3LYP/6-311++G(d,p) (LANL2TZ(f) for Pt) single-point calculations with the integral equation formalism polarizable continuum model (IEFPCM) in dichloromethane. The radii and nonelectrostatic terms were taken from Truhlar and co-workers’ universal solvation model (SMD). Three pathways which lead to the formation of allene via a [1,2]-C–C bond migration (regioselectivity) and C–Si bond migration were performed. Our calculations suggest the following. (1) The major pathway of the cycle involves an initial [1,2]-silyl shift leading to a platinum intermediate. A subsequent [1,2]-C–C bond shift of this intermediate then provides the corresponding allenes. (2) Due to the consequence of an interaction between the Lewis acidic platinum and the allene, the rearrangement of α-alkoxy-substituted cyclopropenes does not give the allene as the final product. (3) The calculations suggest that the electronic effect of a silyl substituent on the cyclopropene is essential for this reaction. Furthermore, the silyl groups provide not only the β-cation-stabilizing effect but also a facile migration group for the whole reaction.
Co-reporter:Ran Fang, Lizi Yang and Yongcheng Wang
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 8) pp:2760-2770
Publication Date(Web):28 Feb 2011
DOI:10.1039/C0OB01098G
The mechanisms of gold(III)-catalyzed synthesis of highly substituted furansvia [3,3]-sigmatropic rearrangements and/or [1,2]-acyloxy migration based on propargyl ketones have been investigated using density functional theory calculations at BHandHLYP/6-31G(d,p) (SDD for Au) level of theory. Solvent effects on these reactions were explored using calculations that included a polarizable continuum model (PCM) for the solvent (toluene). Two plausible pathways that lead to the formation of Au(III) vinyl carbenoid and an allenyl structure through [3,3]-sigmatropic rearrangements, [1,2]-acyloxy migration viaoxirenium and dioxolenylium were performed. Our calculated results suggested: (1) the major pathway of the cycle causes an initial Rautenstrauch-type [1,2]-migration viaoxirenium to form an Au(III) vinyl carbenoid. Subsequent cycloisomerization of this intermediate then provides the corresponding furan whether for the methyl-substituted propargylic acetates or the phenyl-substituted propargylic acetates; (2) for the methyl-substituted propargylic acetates, the formation of Au(III) vinyl carbenoid structures was the rate-determining step. However, intramolecular nucleophilic attack and subsequent cycloisomerization to give the final product was rate-determining for the phenyl-substituted propargylic acetates. The computational results are consistent with the experimental observations of Gevorgyan, et al. for gold(III)-catalyzed synthesis of highly substituted furans based on propargyl ketones.
Co-reporter:Xiaoxiao Wei, Ran Fang and Lizi Yang
Catalysis Science & Technology (2011-Present) 2015 - vol. 5(Issue 6) pp:NaN3362-3362
Publication Date(Web):2015/04/20
DOI:10.1039/C5CY00407A
The reaction mechanisms of N-heterocylic carbene (NHC)-catalyzed annulation to 3-pyrancarbaldehydes or cyclopentene were theoretically analyzed. Different from what has been reported for normal aldehydes or enals, our calculation results revealed that the intermediate results from the conjugate addition to the central carbon atom of the allene moiety, instead of forming the Breslow intermediate. The next step, 1,4-addition of the chalcone, resulted in the formation of an enolate anion intermediate. Then, intramolecular conjugate addition and elimination of the NHC catalyst afforded the final product. In addition, the computed results showed good agreement with the experimental evidence, suggesting that both the existence of an allene structure and charge distribution play a crucial role in the outcome of chemoselectivity and involvement of operative intermediates in the reaction pathway.
Co-reporter:Ran Fang, Xiaoxiao Wei and Lizi Yang
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 42) pp:NaN8441-8441
Publication Date(Web):2014/09/01
DOI:10.1039/C4OB00894D
We report here the theoretical analysis of the mechanism and regioselectivity of gold(I) or platinum(II) catalyzed intramolecular hydroarylation to pyrrolopyridinones and pyrroloazepinones. AuPH3+ and PtCl2 have been considered to account for some experimental observations. Our calculation results indicate that in the case of cationic gold the nucleophilic attack of the pyrrole on the activated alkyne occurs in an exo-dig fashion generating a six-membered intermediate, which upon deprotonation and protodeauration forms pyrrolopyridinone. When platinum is used, an endo-dig fashion is observed generating a seven-membered intermediate. After deprotonation and protodeplatination pyrroloazepinone is formed. Whether for exo-dig (gold(I)) or endo-dig (platinum(II)) cyclization, a [1,2]-migration would not be needed.
Co-reporter:Ran Fang, Lizi Yang and Yongcheng Wang
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 8) pp:NaN2770-2770
Publication Date(Web):2011/02/28
DOI:10.1039/C0OB01098G
The mechanisms of gold(III)-catalyzed synthesis of highly substituted furansvia [3,3]-sigmatropic rearrangements and/or [1,2]-acyloxy migration based on propargyl ketones have been investigated using density functional theory calculations at BHandHLYP/6-31G(d,p) (SDD for Au) level of theory. Solvent effects on these reactions were explored using calculations that included a polarizable continuum model (PCM) for the solvent (toluene). Two plausible pathways that lead to the formation of Au(III) vinyl carbenoid and an allenyl structure through [3,3]-sigmatropic rearrangements, [1,2]-acyloxy migration viaoxirenium and dioxolenylium were performed. Our calculated results suggested: (1) the major pathway of the cycle causes an initial Rautenstrauch-type [1,2]-migration viaoxirenium to form an Au(III) vinyl carbenoid. Subsequent cycloisomerization of this intermediate then provides the corresponding furan whether for the methyl-substituted propargylic acetates or the phenyl-substituted propargylic acetates; (2) for the methyl-substituted propargylic acetates, the formation of Au(III) vinyl carbenoid structures was the rate-determining step. However, intramolecular nucleophilic attack and subsequent cycloisomerization to give the final product was rate-determining for the phenyl-substituted propargylic acetates. The computational results are consistent with the experimental observations of Gevorgyan, et al. for gold(III)-catalyzed synthesis of highly substituted furans based on propargyl ketones.



![1H-Imidazole, 1,1'-[1,1'-biphenyl]-4,4'-diylbis-](/data/chemimg/102600/855766-92-6.png)
![1H-Imidazole, 1,1'-[1,1'-biphenyl]-4,4'-diylbis-](/data/chemimg/102600/855766-92-6_b.png)

































![Imidazo[2,1-b]thiazole,3-methyl-6-phenyl-](http://img.cochemist.com/ccimg/26000/25968-20-1.png)
![Imidazo[2,1-b]thiazole,3-methyl-6-phenyl-](http://img.cochemist.com/ccimg/26000/25968-20-1_b.png)
![Imidazo[2,1-b]benzothiazole, 5,6,7,8-tetrahydro-2-phenyl-](http://img.cochemist.com/ccimg/25800/25752-17-4.png)
![Imidazo[2,1-b]benzothiazole, 5,6,7,8-tetrahydro-2-phenyl-](http://img.cochemist.com/ccimg/25800/25752-17-4_b.png)






![2-Phenylbenzo[d]imidazo[2,1-b]thiazole](http://img.cochemist.com/ccimg/17900/17833-07-7.png)
![2-Phenylbenzo[d]imidazo[2,1-b]thiazole](http://img.cochemist.com/ccimg/17900/17833-07-7_b.png)
![6-PHENYLIMIDAZO[2,1-B]THIAZOLE](http://img.cochemist.com/ccimg/7100/7008-63-1.png)
![6-PHENYLIMIDAZO[2,1-B]THIAZOLE](http://img.cochemist.com/ccimg/7100/7008-63-1_b.png)