Co-reporter:Brian P. Woods, Manuel Orlandi, Chung-Yang Huang, Matthew S. Sigman, and Abigail G. Doyle
Journal of the American Chemical Society April 26, 2017 Volume 139(Issue 16) pp:5688-5688
Publication Date(Web):April 13, 2017
DOI:10.1021/jacs.7b03448
A Ni-catalyzed reductive cross-coupling of styrenyl aziridines with aryl iodides is reported. This reaction proceeds by a stereoconvergent mechanism and is thus amenable to asymmetric catalysis using a chiral bioxazoline ligand for Ni. The process allows facile access to highly enantioenriched 2-arylphenethylamines from racemic aziridines. Multivariate analysis revealed that ligand polarizability, among other features, influences the observed enantioselectivity, shedding light on the success of this emerging ligand class for enantioselective Ni catalysis.
Co-reporter:Matthew K. Nielsen;Benjamin J. Shields;Junyi Liu;Dr. Michael J. Williams;Dr. Michael J. Zacuto; Abigail G. Doyle
Angewandte Chemie 2017 Volume 129(Issue 25) pp:7297-7300
Publication Date(Web):2017/06/12
DOI:10.1002/ange.201702079
AbstractWe report a redox-neutral formylation of aryl chlorides that proceeds through selective 2-functionalization of 1,3-dioxolane through nickel and photoredox catalysis. This scalable benchtop approach provides a distinct advantage over traditional reductive carbonylation in that no carbon monoxide, pressurized gas, or stoichiometric reductant is employed. The mild conditions give unprecedented scope from abundant and complex aryl chloride starting materials.
Co-reporter:Matthew K. Nielsen;Benjamin J. Shields;Junyi Liu;Dr. Michael J. Williams;Dr. Michael J. Zacuto; Abigail G. Doyle
Angewandte Chemie International Edition 2017 Volume 56(Issue 25) pp:7191-7194
Publication Date(Web):2017/06/12
DOI:10.1002/anie.201702079
AbstractWe report a redox-neutral formylation of aryl chlorides that proceeds through selective 2-functionalization of 1,3-dioxolane through nickel and photoredox catalysis. This scalable benchtop approach provides a distinct advantage over traditional reductive carbonylation in that no carbon monoxide, pressurized gas, or stoichiometric reductant is employed. The mild conditions give unprecedented scope from abundant and complex aryl chloride starting materials.
Co-reporter:Erin E. Gray, Matthew K. Nielsen, Kimberly A. Choquette, Julia A. Kalow, Thomas J. A. Graham, and Abigail G. Doyle
Journal of the American Chemical Society 2016 Volume 138(Issue 34) pp:10802-10805
Publication Date(Web):August 8, 2016
DOI:10.1021/jacs.6b06770
The copper-catalyzed H–F insertion into α-diazocarbonyl compounds is described using potassium fluoride (KF) and hexafluoroisopropanol. Access to complex α-fluorocarbonyl derivatives is achieved under mild conditions, and the method is readily adapted to radiofluorination with [18F]KF. This late-stage strategy provides an attractive route to 18F-labeled biomolecules.
Co-reporter:Benjamin J. Shields and Abigail G. Doyle
Journal of the American Chemical Society 2016 Volume 138(Issue 39) pp:12719-12722
Publication Date(Web):September 21, 2016
DOI:10.1021/jacs.6b08397
Here we report the development of a C(sp3)–H cross-coupling platform enabled by the catalytic generation of chlorine radicals by nickel and photoredox catalysis. Aryl chlorides serve as both cross-coupling partners and the chlorine radical source for the α-oxy C(sp3)–H arylation of cyclic and acyclic ethers. Mechanistic studies suggest that photolysis of a Ni(III) aryl chloride intermediate, generated by photoredox-mediated single-electron oxidation, leads to elimination of a chlorine radical in what amounts to the sequential capture of two photons. Arylations of a benzylic C(sp3)–H bond of toluene and a completely unactivated C(sp3)–H bond of cyclohexane demonstrate the broad implications of this manifold for accomplishing numerous C(sp3)–H bond functionalizations under exceptionally mild conditions.
Co-reporter:J. Patrick Lutz, Stephen T. Chau and Abigail G. Doyle
Chemical Science 2016 vol. 7(Issue 7) pp:4105-4109
Publication Date(Web):08 Mar 2016
DOI:10.1039/C6SC00702C
We report an enantioselective Ni-catalyzed cross coupling of arylzinc reagents with pyridinium ions formed in situ from pyridine and a chloroformate. This reaction provides enantioenriched 2-aryl-1,2-dihydropyridine products that can be elaborated to numerous piperidine derivatives with little or no loss in ee. This method is notable for its use of pyridine, a feedstock chemical, to build a versatile, chiral heterocycle in a single synthetic step.
Co-reporter:Dr. Cice L. Joe ; Abigail G. Doyle
Angewandte Chemie 2016 Volume 128( Issue 12) pp:4108-4111
Publication Date(Web):
DOI:10.1002/ange.201511438
Abstract
Using nickel and photoredox catalysis, the direct functionalization of C(sp3)−H bonds of N-aryl amines by acyl electrophiles is described. The method affords a diverse range of α-amino ketones at room temperature and is amenable to late-stage coupling of complex and biologically relevant groups. C(sp3)−H activation occurs by photoredox-mediated oxidation to generate α-amino radicals which are intercepted by nickel in catalytic C(sp3)−C coupling. The merger of these two modes of catalysis leverages nickel's unique properties in alkyl cross-coupling while avoiding limitations commonly associated with transition-metal-mediated C(sp3)−H activation, including requirements for chelating directing groups and high reaction temperatures.
Co-reporter:Dr. Cice L. Joe ; Abigail G. Doyle
Angewandte Chemie International Edition 2016 Volume 55( Issue 12) pp:4040-4043
Publication Date(Web):
DOI:10.1002/anie.201511438
Abstract
Using nickel and photoredox catalysis, the direct functionalization of C(sp3)−H bonds of N-aryl amines by acyl electrophiles is described. The method affords a diverse range of α-amino ketones at room temperature and is amenable to late-stage coupling of complex and biologically relevant groups. C(sp3)−H activation occurs by photoredox-mediated oxidation to generate α-amino radicals which are intercepted by nickel in catalytic C(sp3)−C coupling. The merger of these two modes of catalysis leverages nickel's unique properties in alkyl cross-coupling while avoiding limitations commonly associated with transition-metal-mediated C(sp3)−H activation, including requirements for chelating directing groups and high reaction temperatures.
Co-reporter:Matthew K. Nielsen; Christian R. Ugaz; Wenping Li
Journal of the American Chemical Society 2015 Volume 137(Issue 30) pp:9571-9574
Publication Date(Web):July 15, 2015
DOI:10.1021/jacs.5b06307
We report an inexpensive, thermally stable deoxyfluorination reagent that fluorinates a broad range of alcohols without substantial formation of elimination side products. This combination of selectivity, safety, and economic viability enables deoxyfluorination on preparatory scale. We employ the [18F]-labeled reagent in the first example of a no-carrier-added deoxy-radiofluorination.
Co-reporter:Chung-Yang (Dennis) Huang
Journal of the American Chemical Society 2015 Volume 137(Issue 17) pp:5638-5641
Publication Date(Web):April 16, 2015
DOI:10.1021/jacs.5b02503
A Ni-catalyzed Negishi cross-coupling with 1,1-disubstituted styrenyl aziridines has been developed. This method delivers valuable β-substituted phenethylamines via a challenging reductive elimination that affords a quaternary carbon. A novel electron-deficient olefin ligand, Fro-DO, proved crucial for achieving high rates and chemoselectivity for C–C bond formation over β-H elimination. This ligand is easy to access, is stable, and presents a modular framework for reaction discovery and optimization. We expect that these attributes, combined with the fact that the ligands impart distinct electronic properties to a metal, will support the invention of new transformations not previously possible using established ligands.
Co-reporter:Jason D. Shields, Erin E. Gray, and Abigail G. Doyle
Organic Letters 2015 Volume 17(Issue 9) pp:2166-2169
Publication Date(Web):April 17, 2015
DOI:10.1021/acs.orglett.5b00766
The synthesis and catalytic activity of [(TMEDA)Ni(o-tolyl)Cl], an air-stable, crystalline solid, is described. This complex is an effective precatalyst in a variety of nickel-catalyzed transformations. The lability of TMEDA allows a wide variety of ligands to be used, including mono- and bidentate phosphines, diimines, and N-heterocyclic carbenes. Preliminary mechanistic studies are also reported, which suggest that [(TMEDA)Ni(o-tolyl)Cl] can activate by either a Ni–B or Ni–Ni transmetalation event, depending on the reaction conditions.
Co-reporter:Kevin M. Arendt ; Abigail G. Doyle
Angewandte Chemie International Edition 2015 Volume 54( Issue 34) pp:9876-9880
Publication Date(Web):
DOI:10.1002/anie.201503936
Abstract
A new substrate class for nickel-catalyzed C(sp3) cross-coupling reactions is reported. α-Oxy radicals generated from benzylic acetals, TMSCl, and a mild reductant can participate in chemoselective cross-coupling with aryl iodides using a 2,6-bis(N-pyrazolyl)pyridine (bpp)/Ni catalyst. The mild, base-free conditions are tolerant of a variety of functional groups on both partners, thus representing an attractive CC bond-forming approach to dialkyl ether synthesis. Characterization of a [(bpp)NiCl] complex relevant to the proposed catalytic cycle is also described.
Co-reporter:Kevin M. Arendt ; Abigail G. Doyle
Angewandte Chemie 2015 Volume 127( Issue 34) pp:10014-10018
Publication Date(Web):
DOI:10.1002/ange.201503936
Abstract
A new substrate class for nickel-catalyzed C(sp3) cross-coupling reactions is reported. α-Oxy radicals generated from benzylic acetals, TMSCl, and a mild reductant can participate in chemoselective cross-coupling with aryl iodides using a 2,6-bis(N-pyrazolyl)pyridine (bpp)/Ni catalyst. The mild, base-free conditions are tolerant of a variety of functional groups on both partners, thus representing an attractive CC bond-forming approach to dialkyl ether synthesis. Characterization of a [(bpp)NiCl] complex relevant to the proposed catalytic cycle is also described.
Co-reporter:Chung-Yang (Dennis) Huang and Abigail G. Doyle
Chemical Reviews 2014 Volume 114(Issue 16) pp:8153
Publication Date(Web):May 28, 2014
DOI:10.1021/cr500036t
Co-reporter:Thomas J. A. Graham ; R. Frederick Lambert ; Karl Ploessl ; Hank F. Kung
Journal of the American Chemical Society 2014 Volume 136(Issue 14) pp:5291-5294
Publication Date(Web):March 17, 2014
DOI:10.1021/ja5025645
Herein, we describe an operationally straightforward radiosynthesis of a chiral transition metal fluoride catalyst, [18F](salen)CoF, and its use for late-stage enantioselective aliphatic radiofluorination. We demonstrate the utility of the method by preparing single enantiomer experimental and clinically validated PET tracers that contain base-sensitive functional groups, epimerizable stereocenters, and nitrogen-rich motifs. Unlike the conventional radiosyntheses of these targets with [18F]KF, labeling with (salen)CoF is possible in the last step and under exceptionally mild conditions. These results constitute a rare example of a nucleophilic radiofluorination using a transition metal fluoride and highlight the potential of such reagents to enhance traditional methods for labeling aliphatic hydrocarbons.
Co-reporter:Jason D. Shields, Derek T. Ahneman, Thomas J. A. Graham, and Abigail G. Doyle
Organic Letters 2014 Volume 16(Issue 1) pp:142-145
Publication Date(Web):November 26, 2013
DOI:10.1021/ol4031364
Quinolinium ions are engaged in an asymmetric, Ni-catalyzed Suzuki cross-coupling to yield 2-aryl- and 2-heteroaryl-1,2-dihydroquinolines. Key to the development of this method is the use of a Ni(II) precatalyst that activates without the need for strong reductants or high temperatures. The Ni–iminium activation mode is demonstrated as an exceptionally mild pathway to generate enantioenriched products from racemic starting materials.
Co-reporter:Zhiwei Zuo;Derek T. Ahneman;Lingling Chu;Jack A. Terrett;David W. C. MacMillan
Science 2014 Volume 345(Issue 6195) pp:437-440
Publication Date(Web):25 Jul 2014
DOI:10.1126/science.1255525
A bright outlook for carbon coupling
In contemporary organic chemistry, it is straightforward to forge bonds between unsaturated carbons (i.e., carbons already engaged in double bonds) using cross-coupling catalysis. The protocol runs into some trouble, however, if one or both starting carbon centers are saturated (purely single-bonded). Tellis et al. and Zuo et al. independently found that combining a second, light-activated catalyst with a nickel cross-coupling catalyst could achieve selective coupling of saturated and unsaturated reagents (see the Perspective by Lloyd-Jones and Ball). Their methods rely on single-electron transfer from the light-activated catalyst to the saturated carbon, thereby enhancing its reactivity more effectively than the twoelectron mechanisms prevailing in traditional protocols.
Science, this issue p. 433, p. 437; see also p. 381
Co-reporter:Matthew H. Katcher, Per-Ola Norrby, and Abigail G. Doyle
Organometallics 2014 Volume 33(Issue 9) pp:2121-2133
Publication Date(Web):January 21, 2014
DOI:10.1021/om401240p
A computational and experimental approach was employed to study the mechanism of the palladium(0)-catalyzed fluorination of allylic chlorides with AgF as fluoride source. Our findings indicate that an allylpalladium fluoride is a key intermediate necessary for the generation of both the nucleophile and electrophile. Evidence was also obtained to support a homobimetallic mechanism in which C–F bond formation occurs by nucleophilic attack of a neutral allylpalladium fluoride on a cationic allylpalladium electrophile (with fluoride as counterion). The high branched selectivity and unusual ligand effects observed in the regioselective fluorination are assessed in light of this mechanism and calculated transition states. These results may have important implications for the mechanism of other transition-metal-catalyzed fluorinations.
Co-reporter:Marie-Gabrielle Braun
Journal of the American Chemical Society 2013 Volume 135(Issue 35) pp:12990-12993
Publication Date(Web):August 15, 2013
DOI:10.1021/ja407223g
The first catalytic allylic C–H fluorination reaction using a nucleophilic fluoride source is reported. Under the influence of a Pd/Cr cocatalyst system, simple olefin substrates undergo fluorination with Et3N·3HF in good yields with high branched:linear regioselectivity. The mild conditions and broad scope make this reaction a powerful alternative to established methods for the preparation of allylic fluorides from prefunctionalized substrates.
Co-reporter:Daniel K. Nielsen ; Chung-Yang (Dennis) Huang
Journal of the American Chemical Society 2013 Volume 135(Issue 36) pp:13605-13609
Publication Date(Web):August 20, 2013
DOI:10.1021/ja4076716
Herein we report a nickel-catalyzed C–C bond-forming reaction between simple alkyl aziridines and organozinc reagents. This method represents the first catalytic cross-coupling reaction employing a nonallylic and nonbenzylic Csp3–N bond as an electrophile. Key to its success is the use of a new N-protecting group (cinsyl or Cn) bearing an electron-deficient olefin that directs oxidative addition and facilitates reductive elimination. Studies pertinent to elucidation of the mechanism of cross coupling are also presented.
Co-reporter:Marie-Gabrielle Braun, Matthew H. Katcher and Abigail G. Doyle
Chemical Science 2013 vol. 4(Issue 3) pp:1216-1220
Publication Date(Web):04 Jan 2013
DOI:10.1039/C2SC22198E
This article demonstrates the first examples of tandem C–C and C–F bond formation for the palladium-catalyzed carbofluorination of allenes. The intramolecular Heck-fluorination cascade provides monofluoromethylated heteroarenes, an important class of products in medicinal chemistry. We also describe an intermolecular variant for the three-component coupling of allenes, aryl iodides, and AgF. Mechanistic studies indicate that C–F bond formation occurs by outer sphere attack of fluoride on an allylpalladium fluoride intermediate.
Co-reporter:Julia A. Kalow, Abigail G. Doyle
Tetrahedron 2013 69(27–28) pp: 5702-5709
Publication Date(Web):
DOI:10.1016/j.tet.2013.01.062
Co-reporter:Stephen T. Chau;J. Patrick Lutz;Kevin Wu ; Abigail G. Doyle
Angewandte Chemie International Edition 2013 Volume 52( Issue 35) pp:9153-9156
Publication Date(Web):
DOI:10.1002/anie.201303994
Co-reporter:Stephen T. Chau;J. Patrick Lutz;Kevin Wu ; Abigail G. Doyle
Angewandte Chemie 2013 Volume 125( Issue 35) pp:9323-9326
Publication Date(Web):
DOI:10.1002/ange.201303994
Co-reporter:Chung-Yang (Dennis) Huang
Journal of the American Chemical Society 2012 Volume 134(Issue 23) pp:9541-9544
Publication Date(Web):March 13, 2012
DOI:10.1021/ja3013825
A nickel-catalyzed cross-coupling reaction between N-sulfonyl aziridines and organozinc reagents is reported. The catalytic system comprises an inexpensive and air-stable Ni(II) source and dimethyl fumarate as ligand. Regioselective synthesis of β-substituted amines is possible under mild and functional-group-tolerant conditions. The stereoselectivity of the reaction is consistent with a stereoconvergent mechanism wherein the sulfonamide directs C–C bond formation.
Co-reporter:Kevin T. Sylvester ; Kevin Wu
Journal of the American Chemical Society 2012 Volume 134(Issue 41) pp:16967-16970
Publication Date(Web):October 2, 2012
DOI:10.1021/ja3079362
The mechanism of a recently reported Suzuki coupling reaction of quinoline-derived allylic N,O-acetals has been studied using a combination of structural, stereochemical, and kinetic isotope effect experiments. The data indicate that C–O activation is facilitated by Lewis acid assistance from the boronic acid coupling partner and an ionic SN1-like mechanism accounts for oxidative addition. In this context, we demonstrate the first direct observation of oxidative addition to a quinolinium salt. Notably, this mechanism is distinct from the more commonly described SN2(′)-type oxidative addition of low-valent transition metals to most allylic electrophiles.
Co-reporter:Thomas J. A. Graham and Abigail G. Doyle
Organic Letters 2012 Volume 14(Issue 6) pp:1616-1619
Publication Date(Web):March 2, 2012
DOI:10.1021/ol300364s
A modular and highly efficient protocol for the synthesis of 2-aryl- and heteroaryl-2H-chromenes is described. Under base-free conditions, readily accessible 2-ethoxy-2H-chromenes undergo Csp3–O activation and Csp3–C bond formation in the presence of an inexpensive nickel catalyst and boronic acids. This new strategy enables broad access to 2-substituted-2H-chromenes and has been applied to the late-stage incorporation of complex molecules, including the pharmaceuticals loratidine and indomethacin methyl ester.
Co-reporter:Julia A. Kalow, Dana E. Schmitt, and Abigail G. Doyle
The Journal of Organic Chemistry 2012 Volume 77(Issue 8) pp:4177-4183
Publication Date(Web):April 10, 2012
DOI:10.1021/jo300433a
Lewis base catalysis promotes the in situ generation of amine-HF reagents from benzoyl fluoride and a non-nucleophilic alcohol. The hydrofluorination of aziridines to provide β-fluoroamines using this latent HF source is described. This protocol displays a broad scope with respect to aziridine substitution and N-protecting groups. Examples of regio- and diastereoselective ring opening to access medicinally relevant β-fluoroamine building blocks are presented.
Co-reporter:Julia A. Kalow
Journal of the American Chemical Society 2011 Volume 133(Issue 40) pp:16001-16012
Publication Date(Web):August 24, 2011
DOI:10.1021/ja207256s
This report describes mechanistic studies of the (salen)Co- and amine-cocatalyzed enantioselective ring opening of epoxides by fluoride. The kinetics of the reaction, as determined by in situ 19F NMR analysis, are characterized by apparent first-order dependence on (salen)Co. Substituent effects, nonlinear effects, and reactivity with a linked (salen)Co catalyst provide evidence for a rate-limiting, bimetallic ring-opening step. To account for these divergent data, we propose a mechanism wherein the active nucleophilic fluorine species is a cobalt fluoride that forms a resting-state dimer. Axial ligation of the amine cocatalyst to (salen)Co facilitates dimer dissociation and is the origin of the observed cooperativity. On the basis of these studies, we show that significant improvements in the rates, turnover numbers, and substrate scope of the fluoride ring-opening reactions can be realized through the use of a linked salen framework. Application of this catalyst system to a rapid (5 min) fluorination to generate the unlabeled analog of a known PET tracer, F-MISO, is reported.
Co-reporter:Matthew H. Katcher ; Allen Sha
Journal of the American Chemical Society 2011 Volume 133(Issue 40) pp:15902-15905
Publication Date(Web):September 6, 2011
DOI:10.1021/ja206960k
This report describes the Pd(0)-catalyzed fluorination of linear allylic chlorides and bromides, yielding branched allylic fluorides in high selectivity. Many of the significant synthetic limitations previously associated with the preparation of these products are overcome by this catalytic method. We also demonstrate that a chiral bisphosphine-ligated palladium catalyst enables highly enantioselective access to a class of branched allylic fluorides that can be readily diversified to valuable fluorinated products.
Co-reporter:Thomas J. A. Graham, Jason D. Shields and Abigail G. Doyle
Chemical Science 2011 vol. 2(Issue 5) pp:980-984
Publication Date(Web):21 Feb 2011
DOI:10.1039/C1SC00026H
A Ni(0) catalyst facilitates efficient C–C bond formation between a wide range of π-neutral and π-deficient aryl boronic acids and N-acyliminium precursors derived from quinoline and isoquinoline. The reaction proceeds under mild conditions and is tolerant of common organic functional groups. In addition, a simple one-pot protocol amenable to the direct use of substituted quinolines is described. Consistent with preliminary data revealing a mild, organoboronate-mediated racemization of enantioenriched N-acyliminium precursors, initial results indicate that a chiral phosphoramidite-ligated nickel catalyst promotes enantioselective arylation of racemic substrate with no evidence of a kinetic resolution during catalysis.
Co-reporter:Daniel K. Nielsen ; Abigail G. Doyle
Angewandte Chemie International Edition 2011 Volume 50( Issue 27) pp:6056-6059
Publication Date(Web):
DOI:10.1002/anie.201101191
Co-reporter:Daniel K. Nielsen ; Abigail G. Doyle
Angewandte Chemie 2011 Volume 123( Issue 27) pp:6180-6183
Publication Date(Web):
DOI:10.1002/ange.201101191
Co-reporter:Julia A. Kalow
Journal of the American Chemical Society 2010 Volume 132(Issue 10) pp:3268-3269
Publication Date(Web):February 17, 2010
DOI:10.1021/ja100161d
An enantioselective method for the synthesis of β-fluoroalcohols by catalytic nucleophilic fluorination of epoxides is described. Mild reaction conditions and high selectivity are made possible by the use of benzoyl fluoride as a soluble, latent source of fluoride anion. A chiral amine and chiral Lewis acid serve as cooperative catalysts for desymmetrizations of five- through eight-membered cyclic epoxides, affording products in up to 95% ee. The cocatalytic protocol is also effective for kinetic resolutions of racemic terminal epoxides, which proceed with krel values as high as 300.
Co-reporter:Derek T. Ahneman and Abigail G. Doyle
Chemical Science (2010-Present) 2016 - vol. 7(Issue 12) pp:NaN7006-7006
Publication Date(Web):2016/07/28
DOI:10.1039/C6SC02815B
We describe the functionalization of α-amino C–H bonds with aryl halides using a combination of nickel and photoredox catalysis. This direct C–H, C–X coupling uses inexpensive and readily available starting materials to generate benzylic amines, an important class of bioactive molecules. Mechanistically, this method features the direct arylation of α-amino radicals mediated by a nickel catalyst. This reactivity is demonstrated for a range of aryl halides and N-aryl amines, with orthogonal scope to existing C–H activation and photoredox methodologies. We also report reactions with several complex aryl halides, demonstrating the potential utility of this approach in late-stage functionalization.
Co-reporter:J. Patrick Lutz, Stephen T. Chau and Abigail G. Doyle
Chemical Science (2010-Present) 2016 - vol. 7(Issue 7) pp:
Publication Date(Web):
DOI:10.1039/C6SC00702C
Co-reporter:Marie-Gabrielle Braun, Matthew H. Katcher and Abigail G. Doyle
Chemical Science (2010-Present) 2013 - vol. 4(Issue 3) pp:NaN1220-1220
Publication Date(Web):2013/01/04
DOI:10.1039/C2SC22198E
This article demonstrates the first examples of tandem C–C and C–F bond formation for the palladium-catalyzed carbofluorination of allenes. The intramolecular Heck-fluorination cascade provides monofluoromethylated heteroarenes, an important class of products in medicinal chemistry. We also describe an intermolecular variant for the three-component coupling of allenes, aryl iodides, and AgF. Mechanistic studies indicate that C–F bond formation occurs by outer sphere attack of fluoride on an allylpalladium fluoride intermediate.
Co-reporter:Thomas J. A. Graham, Jason D. Shields and Abigail G. Doyle
Chemical Science (2010-Present) 2011 - vol. 2(Issue 5) pp:NaN984-984
Publication Date(Web):2011/02/21
DOI:10.1039/C1SC00026H
A Ni(0) catalyst facilitates efficient C–C bond formation between a wide range of π-neutral and π-deficient aryl boronic acids and N-acyliminium precursors derived from quinoline and isoquinoline. The reaction proceeds under mild conditions and is tolerant of common organic functional groups. In addition, a simple one-pot protocol amenable to the direct use of substituted quinolines is described. Consistent with preliminary data revealing a mild, organoboronate-mediated racemization of enantioenriched N-acyliminium precursors, initial results indicate that a chiral phosphoramidite-ligated nickel catalyst promotes enantioselective arylation of racemic substrate with no evidence of a kinetic resolution during catalysis.
.jpg)
![Dichloro[2-(4,5-dihydro-2-oxazolyl)quinoline]palladium(II)](http://img.cochemist.com/ccimg/1150100/1150097-98-5.png)
![Dichloro[2-(4,5-dihydro-2-oxazolyl)quinoline]palladium(II)](http://img.cochemist.com/ccimg/1150100/1150097-98-5_b.png)
![Benzene, [[[(2S)-2-fluoro-3-buten-1-yl]oxy]methyl]-](http://img.cochemist.com/ccimg/921100/921038-76-8.png)
![Benzene, [[[(2S)-2-fluoro-3-buten-1-yl]oxy]methyl]-](http://img.cochemist.com/ccimg/921100/921038-76-8_b.png)
![Aziridine, 1-[(4-methylphenyl)sulfonyl]-2-(4-penten-1-yl)-](http://img.cochemist.com/ccimg/918200/918160-54-0.png)
![Aziridine, 1-[(4-methylphenyl)sulfonyl]-2-(4-penten-1-yl)-](http://img.cochemist.com/ccimg/918200/918160-54-0_b.png)
![2-[1-(4-METHYLPHENYL)SULFONYLAZIRIDIN-2-YL]PYRIDINE](http://img.cochemist.com/ccimg/797000/796975-18-3.png)
![2-[1-(4-METHYLPHENYL)SULFONYLAZIRIDIN-2-YL]PYRIDINE](http://img.cochemist.com/ccimg/797000/796975-18-3_b.png)
![Benzene, [[[(2E)-4-chloro-2-butenyl]oxy]methyl]-](http://img.cochemist.com/ccimg/689300/689292-42-0.png)
![Benzene, [[[(2E)-4-chloro-2-butenyl]oxy]methyl]-](http://img.cochemist.com/ccimg/689300/689292-42-0_b.png)
![Benzenemethanamine, N-[(1R,2R)-2-fluorocyclohexyl]-, rel-](http://img.cochemist.com/ccimg/654700/654677-27-7.png)
![Benzenemethanamine, N-[(1R,2R)-2-fluorocyclohexyl]-, rel-](http://img.cochemist.com/ccimg/654700/654677-27-7_b.png)
![Benzenesulfonamide, N-[(1R,2R)-2-fluorocyclohexyl]-4-methyl-, rel-](http://img.cochemist.com/ccimg/654700/654676-64-9.png)
![Benzenesulfonamide, N-[(1R,2R)-2-fluorocyclohexyl]-4-methyl-, rel-](http://img.cochemist.com/ccimg/654700/654676-64-9_b.png)
![CARBAMIC ACID, [(2R)-OXIRANYLMETHYL]-, PHENYLMETHYL ESTER](http://img.cochemist.com/ccimg/641700/641616-99-1.png)
![CARBAMIC ACID, [(2R)-OXIRANYLMETHYL]-, PHENYLMETHYL ESTER](http://img.cochemist.com/ccimg/641700/641616-99-1_b.png)





![7-Azabicyclo[4.1.0]heptane, 7-(2-methoxyphenyl)-](http://img.cochemist.com/ccimg/496900/496816-69-4.png)
![7-Azabicyclo[4.1.0]heptane, 7-(2-methoxyphenyl)-](http://img.cochemist.com/ccimg/496900/496816-69-4_b.png)
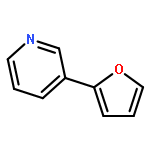
![7-Azabicyclo[4.1.0]heptane, 7-(2-pyridinyl)-](http://img.cochemist.com/ccimg/496900/496816-66-1.png)
![7-Azabicyclo[4.1.0]heptane, 7-(2-pyridinyl)-](http://img.cochemist.com/ccimg/496900/496816-66-1_b.png)
![Aziridine, 1-[(4-methylphenyl)sulfonyl]-2-(2-propenyl)-](http://img.cochemist.com/ccimg/404400/404362-16-9.png)
![Aziridine, 1-[(4-methylphenyl)sulfonyl]-2-(2-propenyl)-](http://img.cochemist.com/ccimg/404400/404362-16-9_b.png)



![Aziridine, 1-[(4-methylphenyl)sulfonyl]-2-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/250300/250260-27-6.png)
![Aziridine, 1-[(4-methylphenyl)sulfonyl]-2-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/250300/250260-27-6_b.png)
![Aziridine, 2-[4-(acetyloxy)phenyl]-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/250300/250260-26-5.png)
![Aziridine, 2-[4-(acetyloxy)phenyl]-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/250300/250260-26-5_b.png)
![Aziridine, 2-(4-fluorophenyl)-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/250300/250260-25-4.png)
![Aziridine, 2-(4-fluorophenyl)-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/250300/250260-25-4_b.png)
![Aziridine, 1-[(1,1-dimethylethyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/238800/238747-35-8.png)
![Aziridine, 1-[(1,1-dimethylethyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/238800/238747-35-8_b.png)


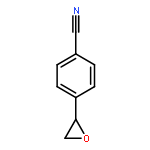
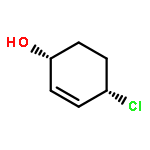
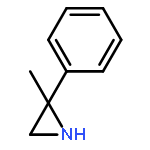
![7-Azabicyclo[4.1.0]heptane, 7-[(1S)-1-phenylethyl]-](http://img.cochemist.com/ccimg/212100/212071-15-3.png)
![7-Azabicyclo[4.1.0]heptane, 7-[(1S)-1-phenylethyl]-](http://img.cochemist.com/ccimg/212100/212071-15-3_b.png)




![Benzenesulfonamide, 4-methyl-N-[1-(phenylmethyl)-3-butenyl]-](http://img.cochemist.com/ccimg/188000/187974-82-9.png)
![Benzenesulfonamide, 4-methyl-N-[1-(phenylmethyl)-3-butenyl]-](http://img.cochemist.com/ccimg/188000/187974-82-9_b.png)
![N,N,2,6-Tetramethyldinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin-4-amine](http://img.cochemist.com/ccimg/185500/185449-86-9.png)
![N,N,2,6-Tetramethyldinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin-4-amine](http://img.cochemist.com/ccimg/185500/185449-86-9_b.png)



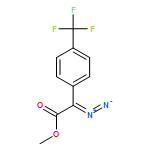
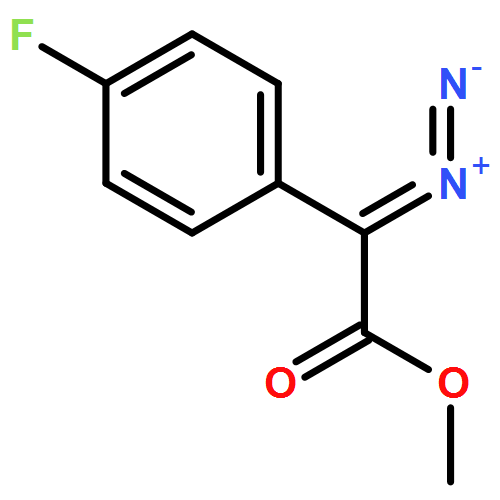
![Aziridine, 1-[(4-nitrophenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/175300/175222-83-0.png)
![Aziridine, 1-[(4-nitrophenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/175300/175222-83-0_b.png)

![Bis[3,5-bis(trifluoromethyl)phenyl]phosphane](http://img.cochemist.com/ccimg/166200/166172-69-6.png)
![Bis[3,5-bis(trifluoromethyl)phenyl]phosphane](http://img.cochemist.com/ccimg/166200/166172-69-6_b.png)
![Tert-butyl 7-azabicyclo[4.1.0]heptane-7-carboxylate](http://img.cochemist.com/ccimg/153800/153789-13-0.png)
![Tert-butyl 7-azabicyclo[4.1.0]heptane-7-carboxylate](http://img.cochemist.com/ccimg/153800/153789-13-0_b.png)
![Benzenesulfinamide,4-methyl-N-(phenylmethylene)-, [S(S)]-](http://img.cochemist.com/ccimg/153300/153277-49-7.png)
![Benzenesulfinamide,4-methyl-N-(phenylmethylene)-, [S(S)]-](http://img.cochemist.com/ccimg/153300/153277-49-7_b.png)


![Aziridine, 1-[(4-methylphenyl)sulfonyl]-2-phenyl-, (2S)-](http://img.cochemist.com/ccimg/149800/149769-84-6.png)
![Aziridine, 1-[(4-methylphenyl)sulfonyl]-2-phenyl-, (2S)-](http://img.cochemist.com/ccimg/149800/149769-84-6_b.png)
![2,5-Pyrrolidinedione, 1-[(diazoacetyl)oxy]-](http://img.cochemist.com/ccimg/147700/147666-20-4.png)
![2,5-Pyrrolidinedione, 1-[(diazoacetyl)oxy]-](http://img.cochemist.com/ccimg/147700/147666-20-4_b.png)
![Zinc, chloro[4-(methoxycarbonyl)phenyl]-](http://img.cochemist.com/ccimg/140500/140483-57-4.png)
![Zinc, chloro[4-(methoxycarbonyl)phenyl]-](http://img.cochemist.com/ccimg/140500/140483-57-4_b.png)


![Aziridine, 2-methyl-1-[(4-methylphenyl)sulfonyl]-3-phenyl-, trans-](http://img.cochemist.com/ccimg/137600/137595-21-2.png)
![Aziridine, 2-methyl-1-[(4-methylphenyl)sulfonyl]-3-phenyl-, trans-](http://img.cochemist.com/ccimg/137600/137595-21-2_b.png)
![2-Octen-1-ol, 8-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (E)-](http://img.cochemist.com/ccimg/136900/136894-04-7.png)
![2-Octen-1-ol, 8-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (E)-](http://img.cochemist.com/ccimg/136900/136894-04-7_b.png)






![Aziridine, 2-butyl-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/117000/116905-61-4.png)
![Aziridine, 2-butyl-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/117000/116905-61-4_b.png)







![Aziridine, 2-(4-bromophenyl)-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/97500/97401-95-1.png)
![Aziridine, 2-(4-bromophenyl)-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/97500/97401-95-1_b.png)
![Aziridine, 2-(4-chlorophenyl)-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/97500/97401-93-9.png)
![Aziridine, 2-(4-chlorophenyl)-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/97500/97401-93-9_b.png)
![Benzene, [[(1E)-1,3-butadienyloxy]methyl]-](http://img.cochemist.com/ccimg/95900/95832-76-1.png)
![Benzene, [[(1E)-1,3-butadienyloxy]methyl]-](http://img.cochemist.com/ccimg/95900/95832-76-1_b.png)
![6-Azabicyclo[3.1.0]hexane, 6-(phenylmethyl)-](http://img.cochemist.com/ccimg/95700/95628-94-7.png)
![6-Azabicyclo[3.1.0]hexane, 6-(phenylmethyl)-](http://img.cochemist.com/ccimg/95700/95628-94-7_b.png)
![Estra-1,3,5(10)-trien-17-one, 3-[[(trifluoromethyl)sulfonyl]oxy]-](http://img.cochemist.com/ccimg/92900/92817-04-4.png)
![Estra-1,3,5(10)-trien-17-one, 3-[[(trifluoromethyl)sulfonyl]oxy]-](http://img.cochemist.com/ccimg/92900/92817-04-4_b.png)

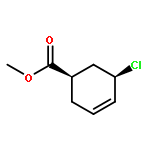
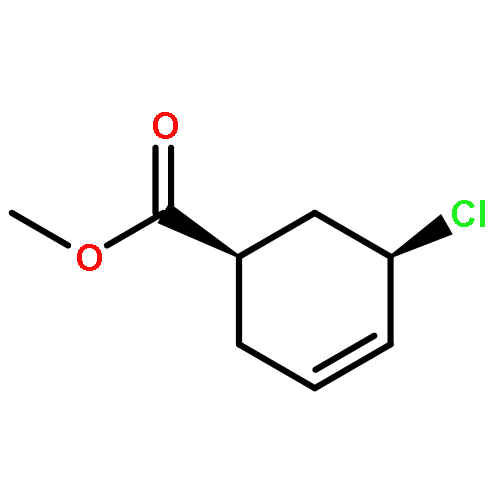
![trans-Methyl 6-oxabicyclo[3.1.0]hexane-3-carboxylate](http://img.cochemist.com/ccimg/86900/86885-57-6.png)
![trans-Methyl 6-oxabicyclo[3.1.0]hexane-3-carboxylate](http://img.cochemist.com/ccimg/86900/86885-57-6_b.png)
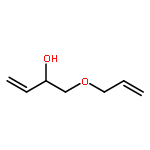
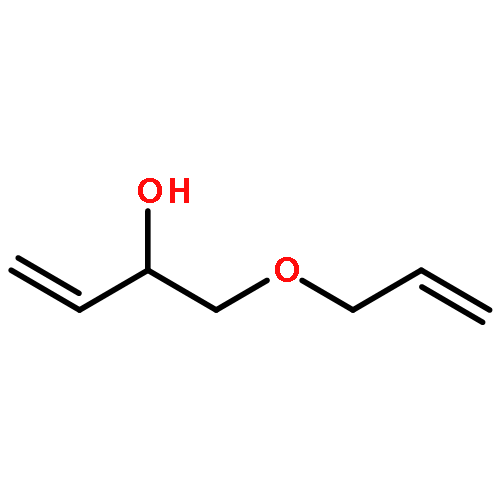
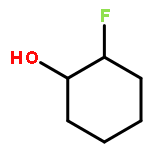
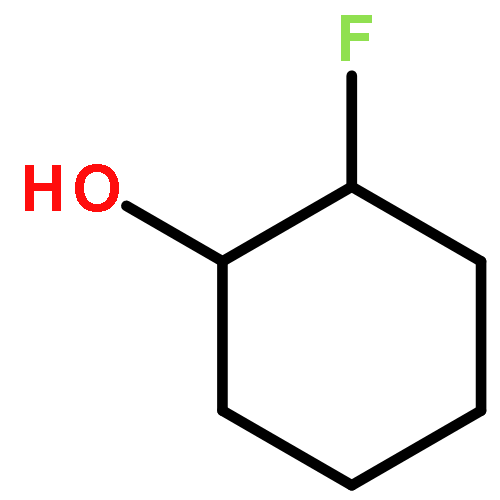
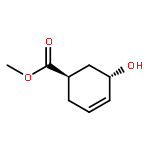
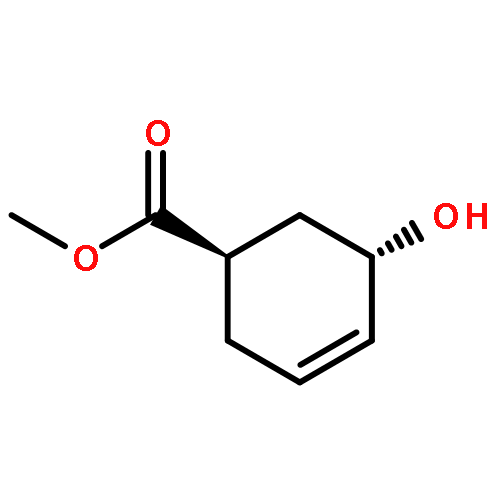







![ETHANONE, 1-[4-(PHENOXYMETHYL)PHENYL]-](http://img.cochemist.com/ccimg/70700/70628-23-8.png)
![ETHANONE, 1-[4-(PHENOXYMETHYL)PHENYL]-](http://img.cochemist.com/ccimg/70700/70628-23-8_b.png)

![7-Azabicyclo[4.1.0]heptane, 7-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/68900/68820-12-2.png)
![7-Azabicyclo[4.1.0]heptane, 7-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/68900/68820-12-2_b.png)


![1H-Indole, 3-ethenyl-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/65100/65037-62-9.png)
![1H-Indole, 3-ethenyl-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/65100/65037-62-9_b.png)
![Aziridine, 1-[(4-methylphenyl)sulfonyl]-2-phenyl-, (R)-](http://img.cochemist.com/ccimg/62600/62596-62-7.png)
![Aziridine, 1-[(4-methylphenyl)sulfonyl]-2-phenyl-, (R)-](http://img.cochemist.com/ccimg/62600/62596-62-7_b.png)


![Cyclohexane, [(1E)-3-chloro-1-propenyl]-](http://img.cochemist.com/ccimg/58700/58649-21-1.png)
![Cyclohexane, [(1E)-3-chloro-1-propenyl]-](http://img.cochemist.com/ccimg/58700/58649-21-1_b.png)







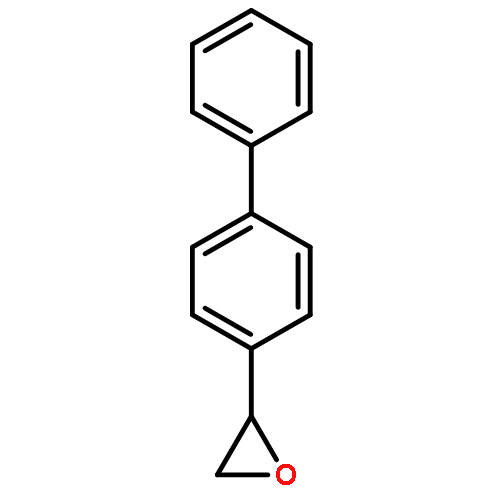

![(3R,8R,9S,10S,12S,13R,14S,17R)-17-((R)-5-(hex-5-en-1-yloxy)-5-oxopentan-2-yl)-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthrene-3,12-diyl diacetate](http://img.cochemist.com/ccimg/33700/33628-48-7.png)
![(3R,8R,9S,10S,12S,13R,14S,17R)-17-((R)-5-(hex-5-en-1-yloxy)-5-oxopentan-2-yl)-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthrene-3,12-diyl diacetate](http://img.cochemist.com/ccimg/33700/33628-48-7_b.png)
![[1,1'-Biphenyl]-4-ethanol,a-phenyl-](http://img.cochemist.com/ccimg/27700/27643-99-8.png)
![[1,1'-Biphenyl]-4-ethanol,a-phenyl-](http://img.cochemist.com/ccimg/27700/27643-99-8_b.png)
![7-Azabicyclo[4.1.0]heptane, 7-(phenylmethyl)-](http://img.cochemist.com/ccimg/24500/24417-01-4.png)
![7-Azabicyclo[4.1.0]heptane, 7-(phenylmethyl)-](http://img.cochemist.com/ccimg/24500/24417-01-4_b.png)
![Aziridine,1-[(4-methylphenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/24400/24395-14-0.png)
![Aziridine,1-[(4-methylphenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/24400/24395-14-0_b.png)



![Aziridine, 1-[(4-methylphenyl)sulfonyl]-2,3-diphenyl-, trans-](http://img.cochemist.com/ccimg/17900/17879-99-1.png)
![Aziridine, 1-[(4-methylphenyl)sulfonyl]-2,3-diphenyl-, trans-](http://img.cochemist.com/ccimg/17900/17879-99-1_b.png)
![Aziridine, 1-[(4-methylphenyl)sulfonyl]-2,3-diphenyl-, cis-](http://img.cochemist.com/ccimg/17900/17879-98-0.png)
![Aziridine, 1-[(4-methylphenyl)sulfonyl]-2,3-diphenyl-, cis-](http://img.cochemist.com/ccimg/17900/17879-98-0_b.png)
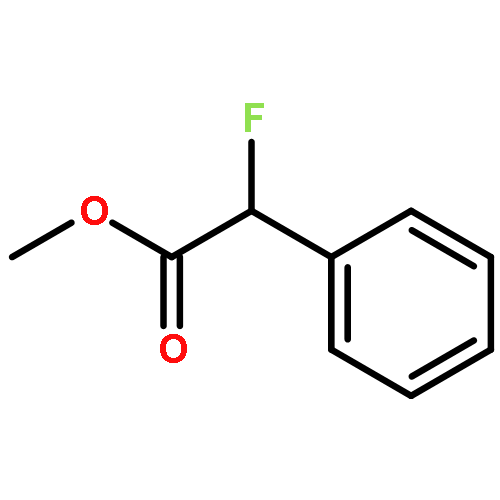
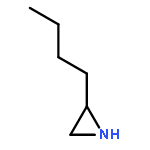
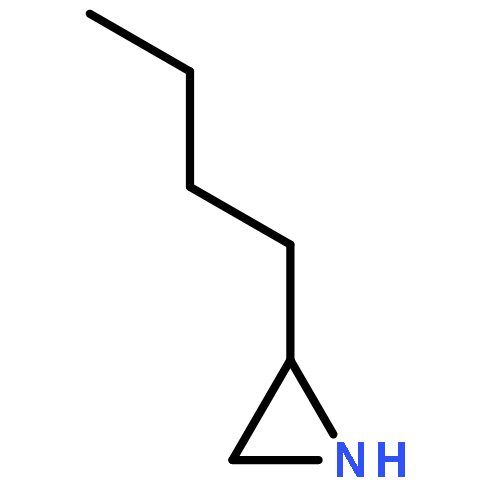


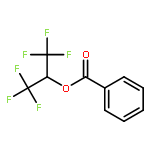


![3-Oxa-6-azabicyclo[3.1.0]hexane](http://img.cochemist.com/ccimg/5900/5834-36-6.png)
![3-Oxa-6-azabicyclo[3.1.0]hexane](http://img.cochemist.com/ccimg/5900/5834-36-6_b.png)


![6-Phenyl-2,3,5,6-tetrahydroimidazo[2,1-b]thiazole](http://img.cochemist.com/ccimg/5100/5036-02-2.png)
![6-Phenyl-2,3,5,6-tetrahydroimidazo[2,1-b]thiazole](http://img.cochemist.com/ccimg/5100/5036-02-2_b.png)
![7-AZABICYCLO[4.1.0]HEPTANE, 7-(4-NITROBENZOYL)-](http://img.cochemist.com/ccimg/4800/4781-44-6.png)
![7-AZABICYCLO[4.1.0]HEPTANE, 7-(4-NITROBENZOYL)-](http://img.cochemist.com/ccimg/4800/4781-44-6_b.png)



![Oxirane, [4-(1,1-dimethylethyl)phenyl]-](http://img.cochemist.com/ccimg/2800/2783-28-0.png)
![Oxirane, [4-(1,1-dimethylethyl)phenyl]-](http://img.cochemist.com/ccimg/2800/2783-28-0_b.png)


![Boroxin,tris[3-(trifluoromethyl)phenyl]- (9CI)](http://img.cochemist.com/ccimg/2300/2265-38-5.png)
![Boroxin,tris[3-(trifluoromethyl)phenyl]- (9CI)](http://img.cochemist.com/ccimg/2300/2265-38-5_b.png)








![8-Azabicyclo[5.1.0]octane(6CI,8CI,9CI)](http://img.cochemist.com/ccimg/300/286-44-2.png)
![8-Azabicyclo[5.1.0]octane(6CI,8CI,9CI)](http://img.cochemist.com/ccimg/300/286-44-2_b.png)
![6-Azabicyclo[3.1.0]hexane](http://img.cochemist.com/ccimg/300/285-63-2.png)
![6-Azabicyclo[3.1.0]hexane](http://img.cochemist.com/ccimg/300/285-63-2_b.png)


![Chromium,chloro[[2,2'-[(1R,2R)-1,2-cyclohexanediylbis[(nitrilo-kN)methylidyne]]bis[4,6-bis(1,1-dimethylethyl)phenolato-kO]](2-)]-, (SP-5-13)-](http://img.cochemist.com/ccimg/165000/164931-83-3.png)
![Chromium,chloro[[2,2'-[(1R,2R)-1,2-cyclohexanediylbis[(nitrilo-kN)methylidyne]]bis[4,6-bis(1,1-dimethylethyl)phenolato-kO]](2-)]-, (SP-5-13)-](http://img.cochemist.com/ccimg/165000/164931-83-3_b.png)
![2-[4-(trifluoromethyl)phenyl]oxirane](http://img.cochemist.com/ccimg/112000/111991-14-1.png)
![2-[4-(trifluoromethyl)phenyl]oxirane](http://img.cochemist.com/ccimg/112000/111991-14-1_b.png)
![Benzamide, N-[(1R,2R)-2-fluorocyclohexyl]-, rel-](http://img.cochemist.com/ccimg/79200/79102-43-5.png)
![Benzamide, N-[(1R,2R)-2-fluorocyclohexyl]-, rel-](http://img.cochemist.com/ccimg/79200/79102-43-5_b.png)
![7-Azabicyclo[4.1.0]heptane, 7-benzoyl-](http://img.cochemist.com/ccimg/4800/4714-50-5.png)
![7-Azabicyclo[4.1.0]heptane, 7-benzoyl-](http://img.cochemist.com/ccimg/4800/4714-50-5_b.png)
![7-Azabicyclo[4.1.0]heptane](http://img.cochemist.com/ccimg/300/286-18-0.png)
![7-Azabicyclo[4.1.0]heptane](http://img.cochemist.com/ccimg/300/286-18-0_b.png)


![Zinc, bromo[4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]phenyl]-](http://img.cochemist.com/ccimg/869700/869683-40-9.png)
![Zinc, bromo[4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]phenyl]-](http://img.cochemist.com/ccimg/869700/869683-40-9_b.png)


![7-AZABICYCLO[4.1.0]HEPTANE, 7-[(4-METHYLPHENYL)SULFONYL]-1-PHENYL-](http://img.cochemist.com/ccimg/645500/645403-26-5.png)
![7-AZABICYCLO[4.1.0]HEPTANE, 7-[(4-METHYLPHENYL)SULFONYL]-1-PHENYL-](http://img.cochemist.com/ccimg/645500/645403-26-5_b.png)
![Benzenesulfonamide, 4-methyl-N-[(1E)-2-phenyl-1-propenyl]-](http://img.cochemist.com/ccimg/602400/602302-01-2.png)
![Benzenesulfonamide, 4-methyl-N-[(1E)-2-phenyl-1-propenyl]-](http://img.cochemist.com/ccimg/602400/602302-01-2_b.png)






![Zinc, bromo[3-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/199500/199465-55-9.png)
![Zinc, bromo[3-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/199500/199465-55-9_b.png)




![Aziridine, 2-methyl-1-[(4-methylphenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/137600/137595-22-3.png)
![Aziridine, 2-methyl-1-[(4-methylphenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/137600/137595-22-3_b.png)
![Boroxin, tris[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/128800/128796-45-2.png)
![Boroxin, tris[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/128800/128796-45-2_b.png)












![Threonine, 4-fluoro-N-[(phenylmethoxy)carbonyl]-, methyl ester](http://img.cochemist.com/ccimg/105000/104916-81-6.png)
![Threonine, 4-fluoro-N-[(phenylmethoxy)carbonyl]-, methyl ester](http://img.cochemist.com/ccimg/105000/104916-81-6_b.png)
![Aziridine, 1-[(4-methylphenyl)sulfonyl]-2,2-diphenyl-](http://img.cochemist.com/ccimg/94300/94286-08-5.png)
![Aziridine, 1-[(4-methylphenyl)sulfonyl]-2,2-diphenyl-](http://img.cochemist.com/ccimg/94300/94286-08-5_b.png)





















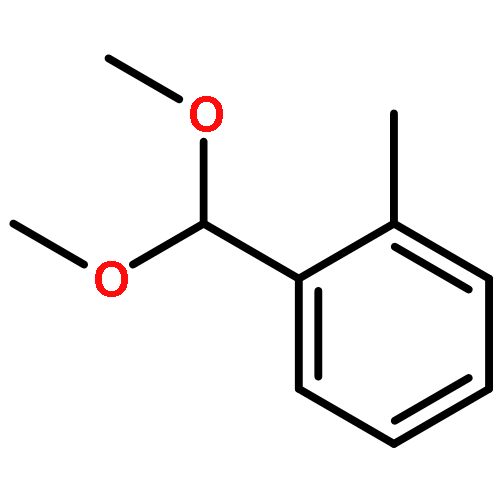




![1-[METHOXY(PHENYL)METHYL]-2-METHYLBENZENE](http://img.cochemist.com/ccimg/55200/55190-63-1.png)
![1-[METHOXY(PHENYL)METHYL]-2-METHYLBENZENE](http://img.cochemist.com/ccimg/55200/55190-63-1_b.png)























![2-Butenedioic acid(2E)-, bis[2,2,2-trifluoro-1-(trifluoromethyl)ethyl] ester (9CI)](http://img.cochemist.com/ccimg/24200/24120-21-6.png)
![2-Butenedioic acid(2E)-, bis[2,2,2-trifluoro-1-(trifluoromethyl)ethyl] ester (9CI)](http://img.cochemist.com/ccimg/24200/24120-21-6_b.png)




![Indeno[1,2-b]azirine, 1,1a,6,6a-tetrahydro-1-[(4-methylphenyl)sulfonyl]-](/data/chemimg/3768100/17798-14-0.png)
![Indeno[1,2-b]azirine, 1,1a,6,6a-tetrahydro-1-[(4-methylphenyl)sulfonyl]-](/data/chemimg/3768100/17798-14-0_b.png)


![Nickel, di-m-chlorobis[(1,2,3-h)-2-methyl-2-propen-1-yl]di-](http://img.cochemist.com/ccimg/12200/12145-60-7.png)
![Nickel, di-m-chlorobis[(1,2,3-h)-2-methyl-2-propen-1-yl]di-](http://img.cochemist.com/ccimg/12200/12145-60-7_b.png)
![Palladium, bis[(1,2,3-h)-2-buten-1-yl]di-m-chlorodi-](http://img.cochemist.com/ccimg/12100/12081-22-0.png)
![Palladium, bis[(1,2,3-h)-2-buten-1-yl]di-m-chlorodi-](http://img.cochemist.com/ccimg/12100/12081-22-0_b.png)










![Oxirane,2-[(2-naphthalenyloxy)methyl]-](http://img.cochemist.com/ccimg/5300/5234-06-0.png)
![Oxirane,2-[(2-naphthalenyloxy)methyl]-](http://img.cochemist.com/ccimg/5300/5234-06-0_b.png)







![Ethanone,1-[1,1'-biphenyl]-3-yl-](http://img.cochemist.com/ccimg/3200/3112-01-4.png)
![Ethanone,1-[1,1'-biphenyl]-3-yl-](http://img.cochemist.com/ccimg/3200/3112-01-4_b.png)