Co-reporter:Dilrukshi Hewage, Wenjin Cao, Jong Hyun Kim, Ya Wang, Yang Liu, and Dong-Sheng Yang
The Journal of Physical Chemistry A 2017 Volume 121(Issue 6) pp:
Publication Date(Web):January 26, 2017
DOI:10.1021/acs.jpca.6b12239
The reaction between La atoms and 1,3-butadiene is carried out in a laser-vaporization molecular beam source. Metal–hydrocarbon species with formulas La(CnHn) (n = 2, 4, and 6) and La(CmHm+2) (m = 4 and 6) are observed with time-of-flight mass spectrometry and characterized with mass-analyzed threshold ionization spectroscopy. A lanthanum–benzene complex [La(C6H6)] is formed by 1,3-butadiene addition to lanthanacyclopropene [La(C2H2)] followed by molecular hydrogen elimination. Lanthanacyclopropene is an intermediate generated by the primary reaction between La and 1,3-butadiene. Two other intermediates produced by the La + 1,3-butadiene reaction are La[η4-(1-buten-3-yne)] [La(C4H4)] and 1-lanthanacyclopent-3-ene [La(C4H6)]. The La(benzene) complex exhibits distinctive metal–ligand bonding from that of the three intermediates as shown by the adiabatic ionization energies and ground electron configurations.
Co-reporter:Zhou Yu, Yu Bai, Yanxiang Wang, Yuxuan Liu, Yanli Zhao, Yang Liu, Kening Sun
Chemical Engineering Journal 2017 Volume 311(Volume 311) pp:
Publication Date(Web):1 March 2017
DOI:10.1016/j.cej.2016.11.093
•NS-GN was prepared through a facile one-step doping process.•Doping graphene with S and/or N atoms created more active sites for I3− reduction.•The N and/or S doping enhanced the electrocatalytic activity of graphene.•The DSSC based on NS-GN showed a high power conversion efficiency of 9.40%.Three-dimensional nitrogen and sulfur co-doped graphene networks are synthesized via a simple one-step hydrothermal route. The nitrogen and sulfur co-doping strategy significantly enhances the electrocatalytic activity for triiodide reduction. The dye-sensitized solar cell based on the nitrogen and sulfur co-doped graphene exhibits an efficiency of 9.40%, which is higher than the efficiency of the Pt-based device. The superior electrochemical performance can be attributed to the synergistic effect of sulfur and nitrogen atoms in graphene, the conductive three-dimensional graphene networks with a large surface area and an interconnected porous structure.
Co-reporter:Yang Liu, Yanxiang Wang, Xiaonan Zheng, Ya Wang
Computational Materials Science 2017 Volume 136, Supplement(Volume 136, Supplement) pp:
Publication Date(Web):1 August 2017
DOI:10.1016/j.commatsci.2017.04.029
•Adsorption abilities of N or/and S doped graphene for I2 are evaluated by DFT.•Synergistic effect by N and S co-doping is revealed based on Bader charge analysis.•Charge and spin distributions are key factors for evaluating adsorption activity.•It provides insight into design of high-performance counter electrodes for DSSC.The carbon materials, as one of the Pt-free alternative materials, have been extensively used for the counter electrodes of dye-sensitized solar cells. In this study, a series of functionalized graphenes with doping N or/and S atoms in different configurations are investigated with density functional theory to reveal their different electrocatalytic activity in the process of I2 adsorption. The results show that the adsorption energy and the transferred charge between the substrate and adsorbed I2 on the surface of N/S co-doped graphene are much bigger than that of N or S monodoped graphene. The enhanced adsorption activity of the former essentially results from the synergistic effect by co-doping with N and S atoms in promoting the positive charge density distribution of active centers and increasing the number of active sites. It is also found that the charge and spin distributions features are two reliable benchmarks for evaluating the physical or chemical adsorption activity of materials and guiding the design of high-performance counter electrodes materials for dye-sensitized solar cells.Download high-res image (171KB)Download full-size image
Co-reporter:Willian Hermoso, Yang Liu, and Isaac B. Bersuker
Journal of Chemical Theory and Computation 2014 Volume 10(Issue 10) pp:4377-4388
Publication Date(Web):August 29, 2014
DOI:10.1021/ct500626j
It is shown that in linear molecules the pseudo-Jahn–Teller (PJT) interaction of a Σ or Π term with a Δ term induces a bending instability that is angular dependent, reducing the symmetry of the adiabatic potential energy surface from expected D∞h to D4h and C∞v to C2v or C4v. This spontaneously broken cylindrical symmetry (BCS) emerges from the solution of the vibronic coupling equations of the PJT effect (PJTE) problems (Σ+Δ)⊗w, (Π+Δ)⊗w, (Π+Σ+Δ)⊗w, and (Δ+Δ)⊗w, where w includes linear, quadratic, and fourth order vibronic coupling terms, and it is confirmed by ab initio calculations for a series of triatomic molecules with ground or excited Δ terms. The BCS is due to the angular symmetry of the electronic wave functions of the Δ term, ∼cos 2φ, and ∼sin 2φ, split by the fourth order vibronic coupling, which in overlap with the other symmetry wave functions of the Σ or Π term provides for the periodical symmetry of the added covalency that facilitates the bending. The mechanism of this PJT-induced BCS effect is discussed in detail; the numerical values of the vibronic coupling parameters for the molecules under consideration were estimated by means of combining separate ab initio calculations of some of them with a procedure fitting the analytical expressions to ab initio calculated energy profiles. It is also shown that the bending of linear molecules in Δ states, similar to Π states, is exclusively a PJT (not Renner–Teller) effect. The BCS revealed in this paper illustrates again the predicting power of the PJTE.
Co-reporter:Yang Liu, Wenli Zou, Isaac B. Bersuker
Chemical Physics Letters 2014 Volume 603() pp:18-20
Publication Date(Web):30 May 2014
DOI:10.1016/j.cplett.2014.04.027
•Noncentrosymmetric BNB ground state is determined by MRCI+Q and MRAQCC methods.•Insufficient electronic correlation in MRCI results in centrosymmetric BNB.•Barrier E(D∞h ← C∞v) increases with the level of electronic correlation treatment.•The origin of off-center distortion is due to Pseudo Jahn–Teller effect.Spontaneous symmetry breaking in the ground state of the BNB molecule is revisited by employing a series of high-level multireference methods based on updated SA-CASSCF wavefunctions with larger active space. It supports our previous determination of two equivalent linear-noncentrosymmetric equilibrium configurations. The calculated magnitude of the barrier between them increases with the level of electronic correlation taken into account. The inadequate centrosymmetric structure obtained by MRCI calculations is caused by insufficient account of electronic correlation. The origin of the off-center distortion in BNB, as in many other systems, is due to the Pseudo Jahn–Teller effect.
Co-reporter:Yang Liu, Isaac B. Bersuker, James E. Boggs
Chemical Physics 2013 Volume 417() pp:26-29
Publication Date(Web):16 May 2013
DOI:10.1016/j.chemphys.2013.02.033



![Benzaldehyde, 2-[[[(1,1-dimethylethyl)diphenylsilyl]oxy]methyl]-](http://img.cochemist.com/ccimg/175200/175158-50-6.png)
![Benzaldehyde, 2-[[[(1,1-dimethylethyl)diphenylsilyl]oxy]methyl]-](http://img.cochemist.com/ccimg/175200/175158-50-6_b.png)




![Benzenemethanol, 2-[[[(1,1-dimethylethyl)diphenylsilyl]oxy]methyl]-](http://img.cochemist.com/ccimg/149200/149170-33-2.png)
![Benzenemethanol, 2-[[[(1,1-dimethylethyl)diphenylsilyl]oxy]methyl]-](http://img.cochemist.com/ccimg/149200/149170-33-2_b.png)



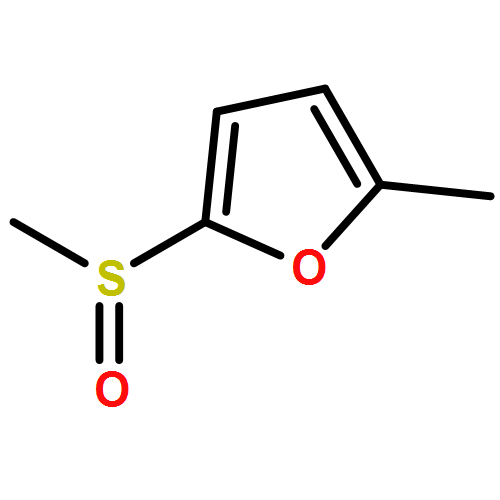


![Phosphonium, [(3-fluorophenyl)methyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/89400/89302-81-8.png)
![Phosphonium, [(3-fluorophenyl)methyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/89400/89302-81-8_b.png)
![Phosphonium, [(2-methoxyphenyl)methyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/64900/64820-07-1.png)
![Phosphonium, [(2-methoxyphenyl)methyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/64900/64820-07-1_b.png)
![Phosphonium, [(2-chlorophenyl)methyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/62700/62640-67-9.png)
![Phosphonium, [(2-chlorophenyl)methyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/62700/62640-67-9_b.png)
![Bicyclo[2.2.1]heptane-2-thione, 1,7,7-trimethyl-, S-oxide, (1R)-](http://img.cochemist.com/ccimg/59200/59121-52-7.png)
![Bicyclo[2.2.1]heptane-2-thione, 1,7,7-trimethyl-, S-oxide, (1R)-](http://img.cochemist.com/ccimg/59200/59121-52-7_b.png)
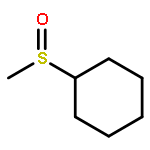
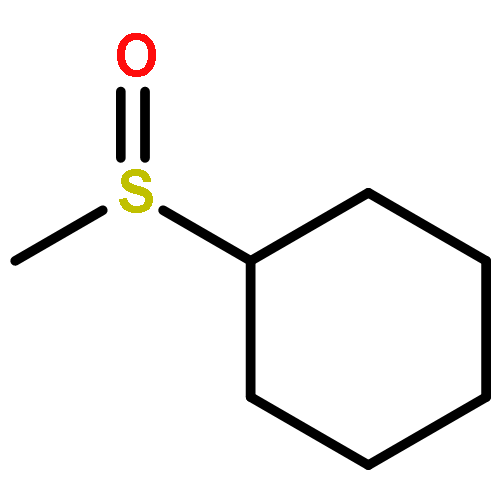


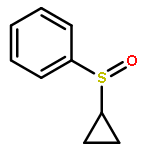




![{[4-(BENZYLOXY)PHENYL]ETHYNYL}(TRIMETHYL)SILANE](http://img.cochemist.com/ccimg/36200/36132-95-3.png)
![{[4-(BENZYLOXY)PHENYL]ETHYNYL}(TRIMETHYL)SILANE](http://img.cochemist.com/ccimg/36200/36132-95-3_b.png)


![Dibenzo[b,e]thiepin-11(6H)-one, 5-oxide](http://img.cochemist.com/ccimg/34200/34129-26-5.png)
![Dibenzo[b,e]thiepin-11(6H)-one, 5-oxide](http://img.cochemist.com/ccimg/34200/34129-26-5_b.png)
![Ethanone, 1-[4-(methylsulfinyl)phenyl]-](http://img.cochemist.com/ccimg/32400/32361-73-2.png)
![Ethanone, 1-[4-(methylsulfinyl)phenyl]-](http://img.cochemist.com/ccimg/32400/32361-73-2_b.png)



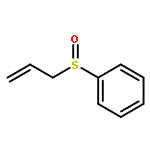
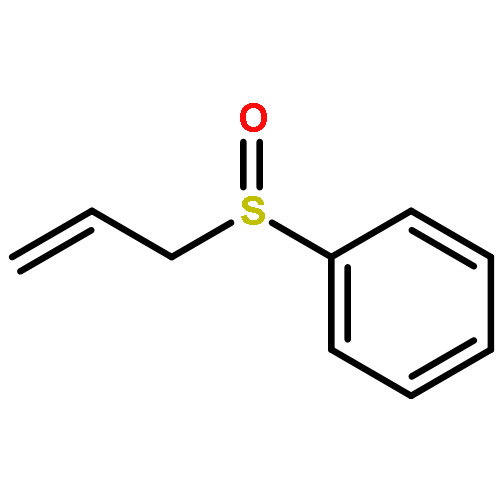
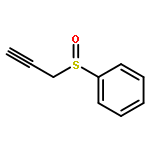
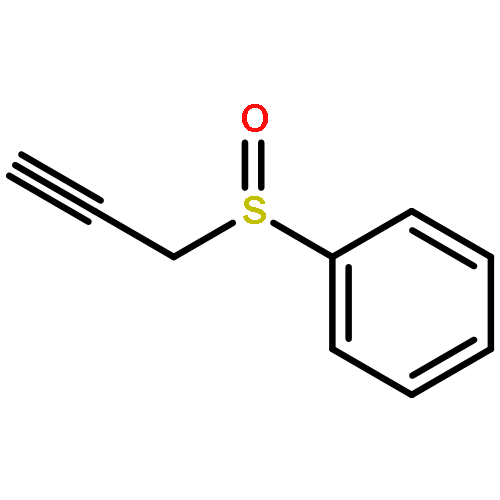




![2,2,4-TRIMETHYL-3-THIABICYCLO[2.2.2]OCTANE](http://img.cochemist.com/ccimg/5800/5718-74-1.png)
![2,2,4-TRIMETHYL-3-THIABICYCLO[2.2.2]OCTANE](http://img.cochemist.com/ccimg/5800/5718-74-1_b.png)


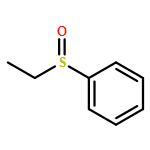
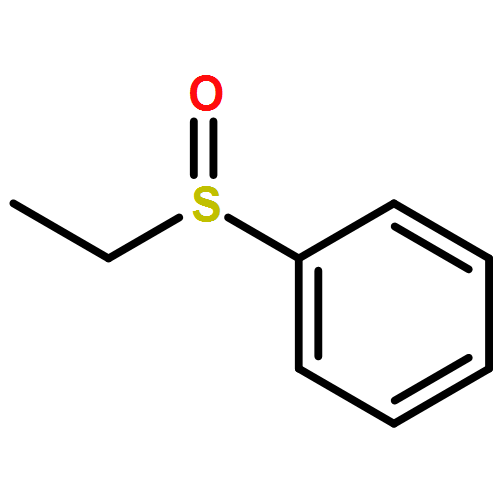
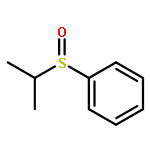
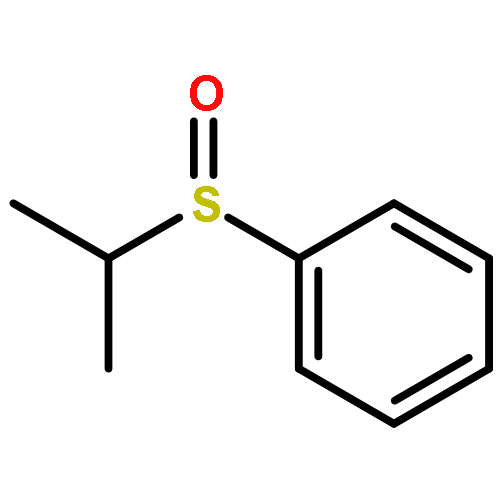

![4-[(1E)-1-{4-[2-(DIMETHYLAMINO)ETHOXY]PHENYL}-2-PHENYL-1-BUTEN-1-<WBR />YL]PHENYL ACETATE](http://img.cochemist.com/ccimg/2400/2394-68-5.png)
![4-[(1E)-1-{4-[2-(DIMETHYLAMINO)ETHOXY]PHENYL}-2-PHENYL-1-BUTEN-1-<WBR />YL]PHENYL ACETATE](http://img.cochemist.com/ccimg/2400/2394-68-5_b.png)
![Phosphonium,[(3-methylphenyl)methyl]triphenyl-, bromide (1:1)](http://img.cochemist.com/ccimg/1800/1702-41-6.png)
![Phosphonium,[(3-methylphenyl)methyl]triphenyl-, bromide (1:1)](http://img.cochemist.com/ccimg/1800/1702-41-6_b.png)



![Benzene, [(phenylmethyl)sulfinyl]-](/data/chemimg/1595300/833-82-9.png)
![Benzene, [(phenylmethyl)sulfinyl]-](/data/chemimg/1595300/833-82-9_b.png)
![Benzene,[(methylsulfinyl)methyl]-](http://img.cochemist.com/ccimg/900/824-86-2.png)
![Benzene,[(methylsulfinyl)methyl]-](http://img.cochemist.com/ccimg/900/824-86-2_b.png)