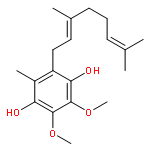
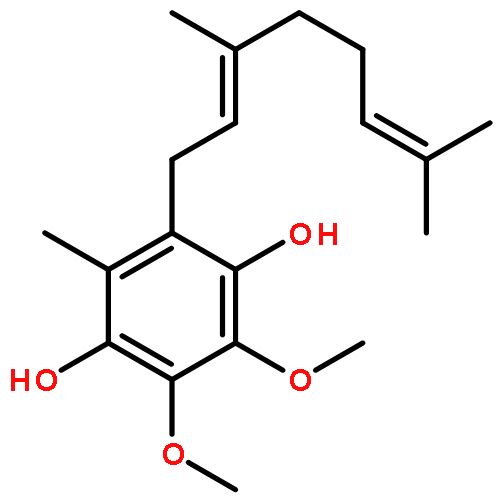
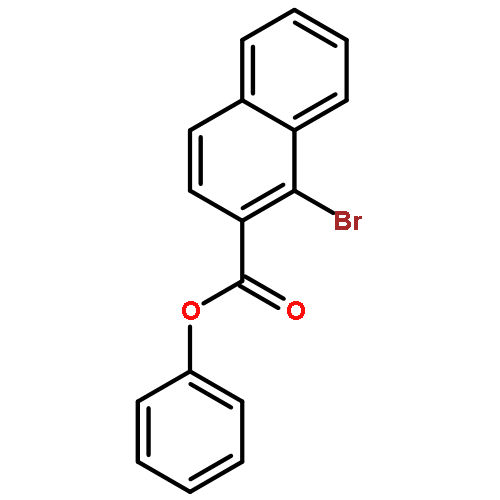
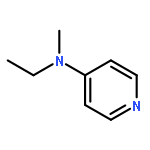
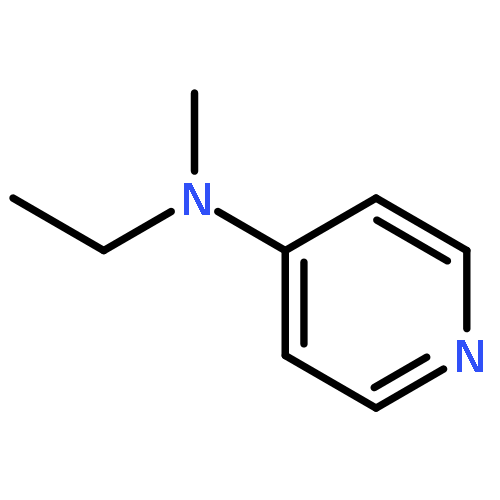
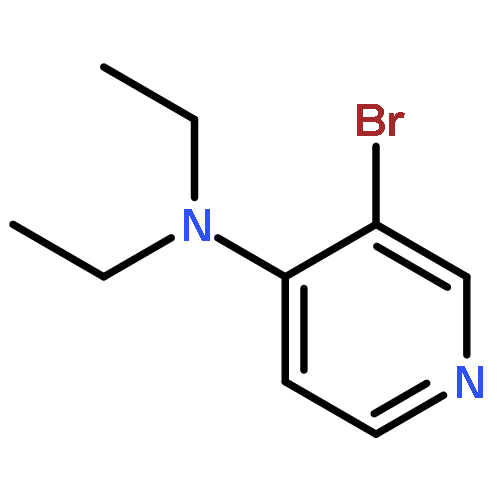
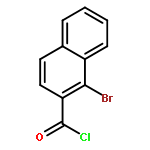
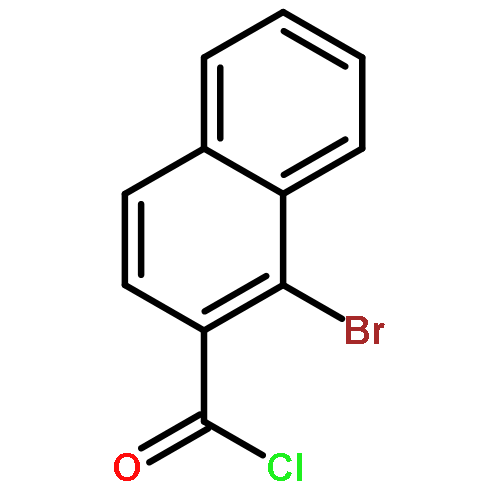
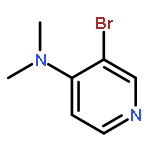
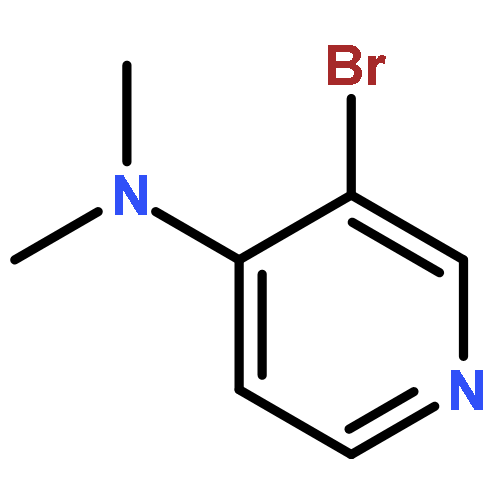
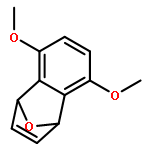
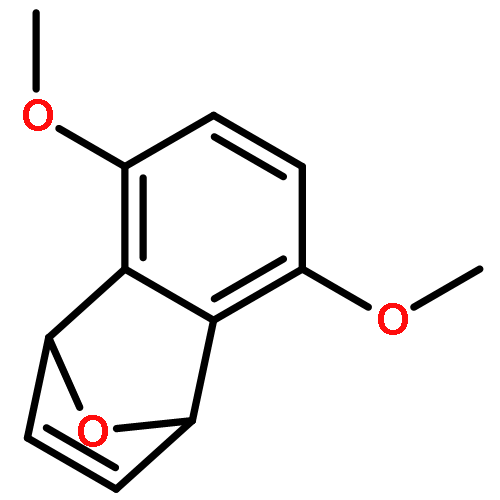
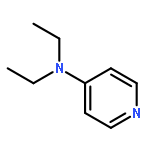
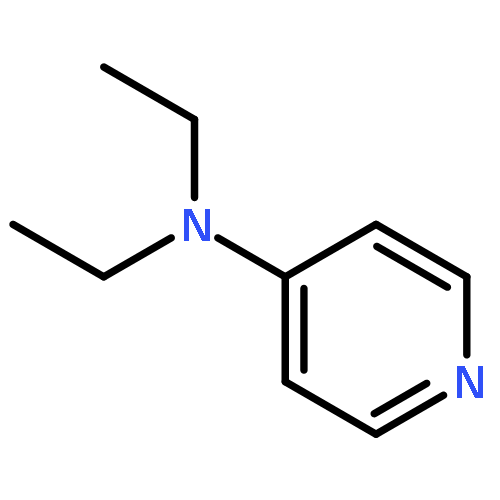
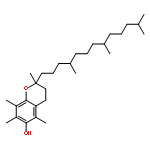
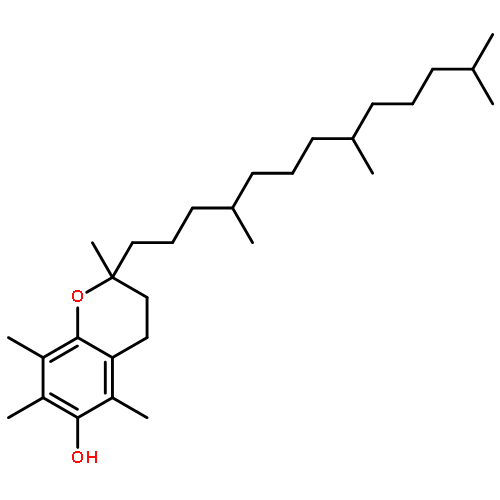
Co-reporter:Takatoshi Fujita, Yohei Haketa, Hiromitsu Maeda, Takeshi Yamamoto
Organic Electronics 2017 Volume 49(Volume 49) pp:
Publication Date(Web):1 October 2017
DOI:10.1016/j.orgel.2017.06.028
•The charge transport in crystal polymorphs of the dipyrrolyldiketone difluoroboron complex were studied.•Utilizing polar diketone difluoroboron complex as the π-conjugated molecule results in the large electronic couplings.•The crystal packing structures have critical effects on the anisotropy and dimensionality of charge-transfer networks.We present a theoretical study on charge transport in crystal polymorphs of the pyrrole-based π-conjugated molecule, the dipyrrolyldiketone difluoroboron complex. The ab initio all-electron fragment molecular orbital calculations were carried out to evaluate electronic couplings for charge transfer in three crystal polymorphs. The electron and hole carrier mobilities were estimated on the basis of the master equation combined with the Marcus theory. We found that utilizing the polar diketone difluoroboron unit as the π-conjugated molecule results in the large molecular interactions and electronic couplings. We demonstrate the impact of crystal packing structures on the anisotropy of charge transport, highlighting the dimensionality of charge-transfer networks varying from zero-dimensional behavior of charge localization to three-dimensional behavior of isotropic charge transport. We further elucidate the effects of different π-π stacking modes on the electronic couplings and intermolecular interactions, which underlie the relationship between the crystal packing structures and charge transport.Download high-res image (171KB)Download full-size image
Co-reporter:Taichi Inagaki and Takeshi Yamamoto
The Journal of Physical Chemistry B 2014 Volume 118(Issue 4) pp:937-950
Publication Date(Web):December 24, 2013
DOI:10.1021/jp410263f
In biomembranes a variety of antioxidants work to suppress oxidative damage. Vitamin E and ubiquinol are among the most important lipid-soluble antioxidants, which trap lipid peroxyl radicals directly or work cooperatively in the regeneration of vitamin E radicals by ubiquinol. Here, we investigate the latter regeneration reaction by using variational transition-state theory with multidimensional tunneling corrections. The result shows that the system forms a compact H-bonded complex by significantly rearranging the donor and acceptor moieties, which leads to a rather narrow potential barrier for H transfer and a very large tunneling effect with a transmission coefficient >4000. In accord with experiment, the Arrhenius activation energy is found to be very small (∼1 kcal/mol), which is interpreted here in terms of mean tunneling energy through the barrier. Regarding the electronic structure, we demonstrate that the present reaction proceeds via a proton-coupled electron transfer (PCET) mechanism and suggest that the PCET character also contributes to the large tunneling effect by sharpening the potential barrier. Finally, a systematic comparison is made among relevant reactions and it is indicated that the antioxidant defense of biomembranes may benefit rather significantly from quantum tunneling to enhance the reaction efficiency.
Co-reporter:Taichi Inagaki, Shinji Aono, Hiroshi Nakano, and Takeshi Yamamoto
The Journal of Physical Chemistry B 2014 Volume 118(Issue 20) pp:5499-5508
Publication Date(Web):May 1, 2014
DOI:10.1021/jp501212y
Despite strong electrostatic repulsion, like-charged ions in aqueous solution can effectively attract each other via ion–water interactions. In this paper we investigate such an effective interaction of like-charged ions in water by using the 3D-RISM-SCF method (i.e., electronic structure theory combined with three-dimensional integral equation theory for molecular solvents). Free energy profiles are calculated at the CCSD(T) level for a series of molecular ions including guanidinium (Gdm+), alkyl-substituted ammonium, and aromatic amine cations. Polarizable continuum model (PCM) and mean-field QM/MM free energy calculations are also performed for comparison. The results show that the stability of like-charged ion pairs in aqueous solution is determined by a very subtle balance between interionic interactions (including dispersion and π-stacking interactions) and ionic solvation/hydrophobic effects and that the Gdm+ ion has a rather favorable character for like-charge association among all the cations studied. Furthermore, we investigate the like-charge pairing in Arg-Ala-Arg and Lys-Ala-Lys tripeptides in water and show that the Arg-Arg pair has a contact free-energy minimum of about −6 kcal/mol. This result indicates that arginine pairing observed on protein surfaces and interfaces is stabilized considerably by solvation effects.
Co-reporter:Hiroshi Nakano and Takeshi Yamamoto
Journal of Chemical Theory and Computation 2013 Volume 9(Issue 1) pp:188-203
Publication Date(Web):November 30, 2012
DOI:10.1021/ct300831t
QM/MM free energy calculation is computationally demanding because of the need for an excessive number of electronic structure calculations. A practical approach for reducing the computational cost is that based on mean field approximation, which calculates the QM wave function in the presence of a partially or totally averaged potential of the MM environment. For obtaining the latter potential, it is common to first represent the QM molecule in terms of point charges and then perform statistical sampling of MM molecules. However, the point charge approximation has the drawback that it tends to overestimate electrostatic (ES) interactions at short-range, which may give rise to a divergence problem in the self-consistent iterations. In this paper, we thus consider a more accurate and robust implementation of mean-field QM/MM method based on continuous QM charge density, here utilizing the following combination: (i) grid-based treatment of ES potential generated by the QM molecule, which allows for an efficient sampling of MM molecules in the presence of QM charge density, and (ii) adaptation of the QM/MM-Ewald method to the mean-field framework for eliminating cutoff errors in the long-range ES interactions. As a numerical test, we apply the obtained method to several benchmark reactions in aqueous solution, and show that the density-based method essentially eliminates the divergence problem while providing the free energy profile consistent with experiment. In addition, we test the utility of a recently proposed screened charge model for the QM charge density and show that the latter also performs well for the free energy calculation. These results suggest that explicit inclusion of charge penetration effects is beneficial for improving the accuracy and stability of the mean-field QM/MM calculation.
Co-reporter:Yusuke Ogihara, Takeshi Yamamoto, and Shigeki Kato
Journal of Chemical Theory and Computation 2011 Volume 7(Issue 8) pp:2507-2519
Publication Date(Web):July 6, 2011
DOI:10.1021/ct200367y
Triplet ketene exhibits a steplike structure in the experimentally observed dissociation rate, but its mechanism is still unclear despite many theoretical efforts. A previous surface-hopping simulation at the CASSCF level suggests that nonadiabatic transition from the S0 to T1 states creates the T1 species in a highly nonstatistical manner, which raises the question of whether the use of statistical rate theory is valid in itself for the T1 state. Here, we study this problem by performing ab initio trajectory simulation at the multireference second-order Möller–Plesset perturbation (MRMP) level of theory. Since the MRMP theory is too expensive for such a trajectory calculation, we first construct dual-level potential energy surfaces (PESs) for the S0 and T1 states by calibrating the PESs at the B3LYP level with a limited set of MRMP energies. We then introduce the assumption of vibrational equilibrium on the S0 surface and characterize the S0 → T1 crossing points using the conditional microcanonical distribution on the S0/T1 seam surface. The latter distribution is obtained by running a constrained trajectory on the seam surface by use of an efficient SHAKE-like method. Subsequently, we propagate a number of T1 trajectories from the seam surface to obtain the dissociation rate. The result shows that (i) the S0 → T1 crossing points are localized mainly in the T1 reactant region; (ii) the lifetime on the T1 surface is about 30 ps at the MRMP level, which is 2 orders of magnitude greater than the previous estimate obtained from the surface-hopping simulation at the CASSCF level (∼100 fs); and (iii) the calculated T1 dissociation rate agrees reasonably well with classical transition state theory. These results suggest that the T1 dissociation is rather statistical, given that the T1 trajectories are initiated from the conditional microcanonical distribution on the seam surface.
Co-reporter:Yusuke Ogihara, Takeshi Yamamoto, Shigeki Kato
Chemical Physics Letters 2011 Volume 511(1–3) pp:28-32
Publication Date(Web):26 July 2011
DOI:10.1016/j.cplett.2011.05.067
Abstract
Triplet ketene exhibits a steplike structure in the experimentally observed photodissociation rates, but its mechanism is still unknown despite many theoretical efforts. Here we revisit this problem by calculating the potential energy profile of triplet ketene with the Adamowicz and Mukherjee multireference coupled-cluster (MRCC) theories. At the MRCCSD level, the imaginary frequency of the dissociation barrier is calculated to be about 300i cm−1, which is slightly smaller than the previous estimates but is still much greater than the expected maximum value for reproducing the observed steps (100i cm−1). This implies that other types of mechanisms (including nonadiabatic ones) may be more plausible for the observed steps.
Co-reporter:Yusuke Ogihara, Takeshi Yamamoto, and Shigeki Kato
The Journal of Physical Chemistry A 2010 Volume 114(Issue 37) pp:9981-9990
Publication Date(Web):August 19, 2010
DOI:10.1021/jp104089m
Triplet ketene exhibits a steplike structure in the experimentally observed dissociation rates, but its mechanism is still unknown despite many theoretical efforts in the past decades. In this paper we revisit this problem by quantum mechanically calculating the reaction probability with multireference-based electronic structure theory. Specifically, we first construct an analytical potential energy surface of triplet state by fitting it to about 6000 ab initio energies computed at the multireference second-order Möller−Plesset perturbation (MRMP2) level. We then evaluate the cumulative reaction probability by using the transition state wave packet method together with an adiabatically constrained Hamiltonian. The result shows that the imaginary barrier frequency on the triplet surface is 328i cm−1, which is close to the CCSD(T) result (321i cm−1) but is likely too large for reproducing the experimentally observed steps. Indeed, our calculated reaction probability exhibits no signature of steps, reflecting too strong tunneling effect along the reaction coordinate. Nevertheless, it is emphasized that the flatness of the potential profile in the transition-state region (which governs the degree of tunneling) depends strongly on the level of electronic structure calculation, thus leaving some possibility that the use of more accurate theories might lead to the observed steps. We also demonstrate that the triplet potential surface differs significantly between the CASSCF and MRMP2 results, particularly in the transition-state region. This fact seems to require more attention when studying the “nonadiabatic” scenario for the steps, in which the crossing seam between S0 and T1 surfaces is assumed to play a central role.
Co-reporter:Takeshi Yamamoto
Chemical Physics Letters 2010 500(4–6) pp: 263-266
Publication Date(Web):
DOI:10.1016/j.cplett.2010.10.014



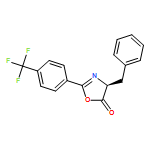
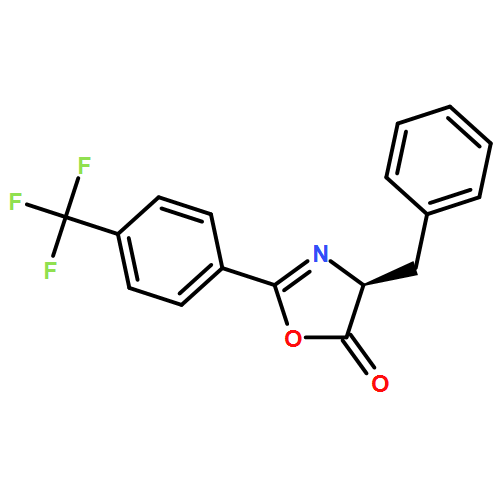


![5(4H)-Oxazolone, 4-methyl-2-[4-(trifluoromethyl)phenyl]-](/data/chemimg/3721500/1361143-04-5.png)
![5(4H)-Oxazolone, 4-methyl-2-[4-(trifluoromethyl)phenyl]-](/data/chemimg/3721500/1361143-04-5_b.png)
![[1,1'-Biphenyl]-2,3-diamine, 2'-(diphenylphosphinyl)-4-methyl-](/data/chemimg/3720700/1134641-49-8.png)
![[1,1'-Biphenyl]-2,3-diamine, 2'-(diphenylphosphinyl)-4-methyl-](/data/chemimg/3720700/1134641-49-8_b.png)
![Silane, trichloro[(1R)-1-phenylethyl]-](http://img.cochemist.com/ccimg/38100/38053-75-7.png)
![Silane, trichloro[(1R)-1-phenylethyl]-](http://img.cochemist.com/ccimg/38100/38053-75-7_b.png)
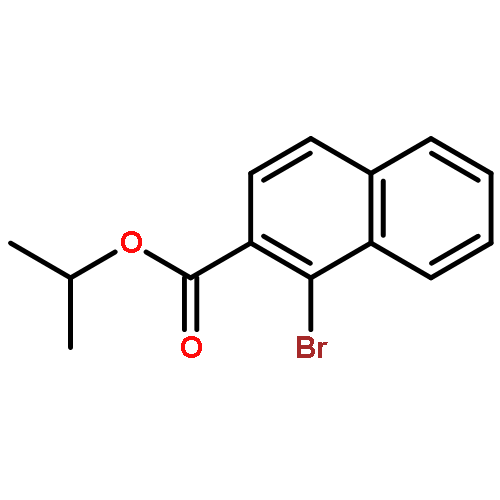
![1H-Naphtho[1,8-de]-1,3,2-diazaborine, 2,3-dihydro-2-phenyl-](http://img.cochemist.com/ccimg/24400/24341-81-9.png)
![1H-Naphtho[1,8-de]-1,3,2-diazaborine, 2,3-dihydro-2-phenyl-](http://img.cochemist.com/ccimg/24400/24341-81-9_b.png)
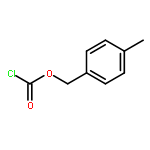

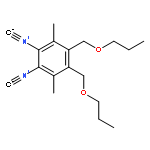
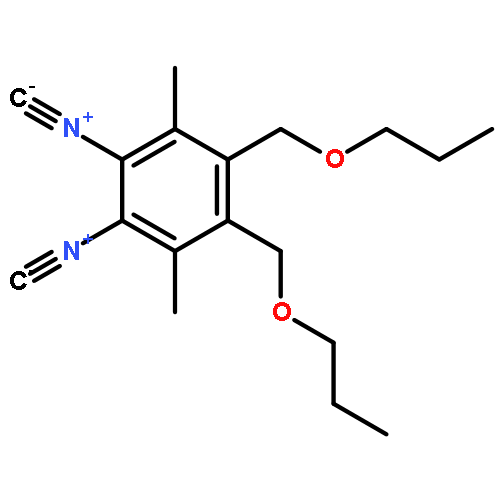




![Silane, trichloro[(1S)-1-phenylethyl]-](http://img.cochemist.com/ccimg/38100/38053-74-6.png)
![Silane, trichloro[(1S)-1-phenylethyl]-](http://img.cochemist.com/ccimg/38100/38053-74-6_b.png)


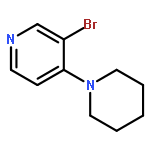
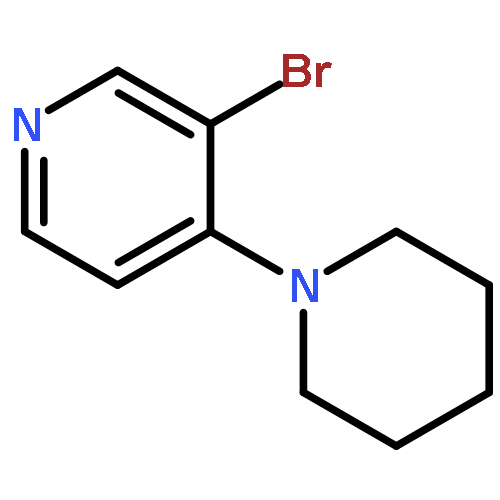
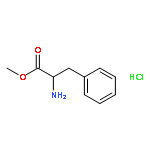
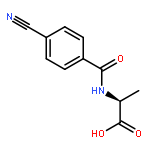
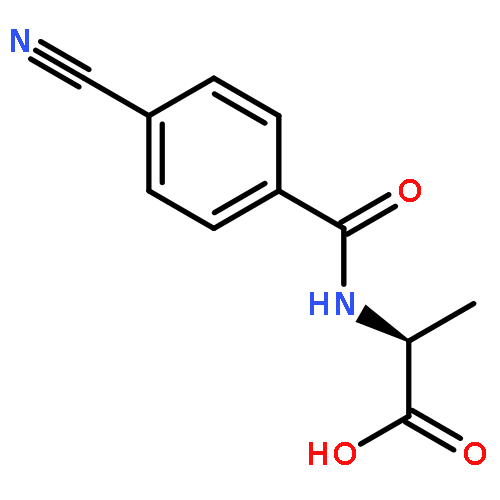
![N-[4-(Trifluoromethyl)benzoyl]-L-methionine methyl ester](http://img.cochemist.com/ccimg/175300/175202-25-2.png)
![N-[4-(Trifluoromethyl)benzoyl]-L-methionine methyl ester](http://img.cochemist.com/ccimg/175300/175202-25-2_b.png)
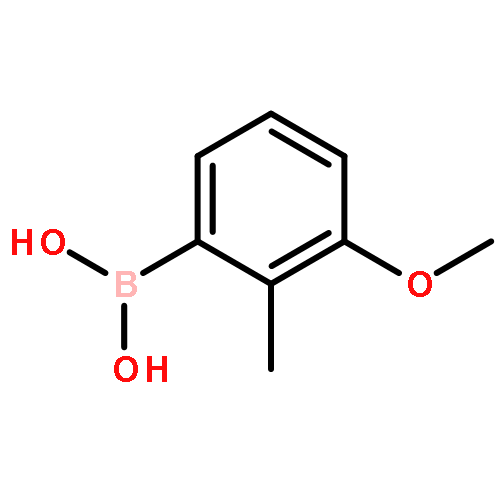
![1,2-bis(4,4,5,5-tetramethyl-[1,3,2]dioxabororan-2-yl)benzene](http://img.cochemist.com/ccimg/269500/269410-07-3.png)
![1,2-bis(4,4,5,5-tetramethyl-[1,3,2]dioxabororan-2-yl)benzene](http://img.cochemist.com/ccimg/269500/269410-07-3_b.png)
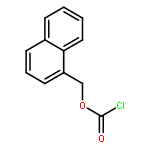
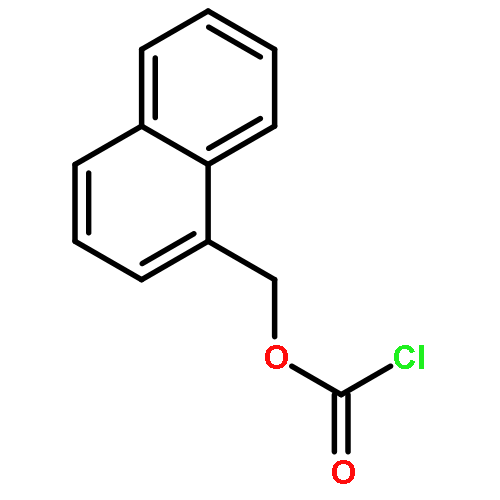




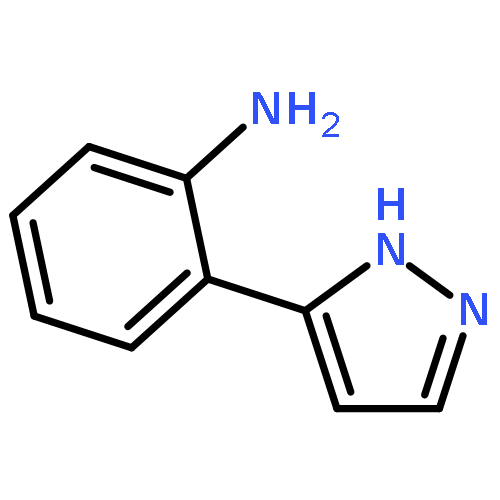
![1-methyl-1,2,3,4-tetrahydro-[1,6]naphthyridine](http://img.cochemist.com/ccimg/636600/636564-06-2.png)
![1-methyl-1,2,3,4-tetrahydro-[1,6]naphthyridine](http://img.cochemist.com/ccimg/636600/636564-06-2_b.png)


![Phosphine, (1R)-[1,1'-binaphthalen]-2-yldiphenyl-](/data/chemimg/1754900/176850-06-9.png)
![Phosphine, (1R)-[1,1'-binaphthalen]-2-yldiphenyl-](/data/chemimg/1754900/176850-06-9_b.png)



![2,3-dihydro-1-methyl-1H-Pyrrolo[3,2-c]pyridine](http://img.cochemist.com/ccimg/219900/219834-79-4.png)
![2,3-dihydro-1-methyl-1H-Pyrrolo[3,2-c]pyridine](http://img.cochemist.com/ccimg/219900/219834-79-4_b.png)