Co-reporter:Jing Wu;Dadian Wang;Xiang Chen;Qingwen Gui;Hua Li;Ze Tan;Guangwei Wang
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 36) pp:7509-7512
Publication Date(Web):2017/09/20
DOI:10.1039/C7OB01726J
A highly efficient synthesis of 4-benzylpyridines was developed via Pd-catalyzed C(sp3)–H arylation between 4-picoline and aryl halides. It was found that the best yields were achieved with a simple Pd(PPh3)4 catalyst and Cs2CO3 as the base. Compared with the known methods, our reaction does not require the use of a strong organometallic reagent as the base.
Co-reporter:Li Wang;Hongyan Zou;Xinwen Zhang
Organic Chemistry Frontiers 2017 vol. 4(Issue 4) pp:587-596
Publication Date(Web):2017/03/28
DOI:10.1039/C6QO00671J
The rhodium-catalyzed intermolecular [3 + 2] cycloadditions of vinylaziridines with allenes represent an efficient method for the synthesis of biologically and pharmaceutically relevant chiral methylene pyrrolidines. In this report, the reaction mechanism was investigated by means of density functional theory calculations. The results show that the Rh–allyl complex was formed through an initial C–N oxidative addition. The subsequent proximal-insertion of allene into the Rh–N bond/C(sp2)–C(sp3) reductive elimination or the distal-insertion of allene into the Rh–C bond led to the formation of 3-methylene-pyrrolidine products or 2-methylene-pyrrolidine products, respectively. The calculations reproduced well the experimentally observed high regio-, E/Z, and diastereo-selectivity. In particular, it was found that the substituent of the allene has a dramatic impact on the proximal-insertion of allene into the Rh–N bond, which enables the selectivity switch between the proximal and distal CC bond insertion upon the change of the substituent. Finally, the high E/Z and diastereo-selectivity is rationalized in terms of the steric repulsion between the allene and the Rh–allyl moiety in the corresponding transition states.
Co-reporter:Genping Huang and Peng Liu
ACS Catalysis 2016 Volume 6(Issue 2) pp:809
Publication Date(Web):December 21, 2015
DOI:10.1021/acscatal.5b02201
The iridium-catalyzed carbonyl-directed hydroarylation of monosubstituted alkenes developed by Bower and co-workers [Crisenza, G. E. M.; McCreanor, N. G.; Bower, J. F. J. Am. Chem. Soc. 2014, 136, 10258–10261] provides an efficient strategy for highly branched-selective hydroarylation of both aryl- and alkyl-substituted alkenes. Density functional theory calculations in the present study revealed that the unique regiochemical control in this reaction is due to an unconventional modified Chalk–Harrod-type mechanism. Instead of the commonly accepted Chalk–Harrod-type mechanism of transition metal-catalyzed hydroarylation that involves C–H oxidative addition, olefin migratory insertion into the Ir–H bond, and C–C reductive elimination, the Ir-catalyzed reaction occurs via migratory insertion of the olefin into the Ir–aryl bond and C–H reductive elimination. The experimentally observed ligand-controlled selectivity is attributed to a combination of electronic and steric effects in the selectivity-determining olefin migratory insertion step. Ligand steric contour maps show that, in reactions with large-bite-angle bisphosphine ligands, such as dFppb, the steric repulsions between the substrate and the aryl substituents on the ligand lead to complete branched selectivity, and the linear selectivity in reactions with small-bite-angle ligands is due to electronic effects that favor 2,1-olefin migratory insertions.Keywords: DFT calculations; hydroarylation; iridium catalyst; mechanism; selectivity
Co-reporter:Mei Zhang and Genping Huang
Dalton Transactions 2016 vol. 45(Issue 8) pp:3552-3557
Publication Date(Web):13 Jan 2016
DOI:10.1039/C5DT04973C
The iridium-catalysed branched-selective hydroarylation of vinyl ethers represents a rare example of the branched-selective hydroarylation involving the non-styrene-type alkenes. Herein, we report our DFT calculations on the mechanism of this reaction. The results show that after C–H oxidative addition, instead of the widely accepted Chalk–Harrod type mechanism, the branched-selective hydroarylation may proceed through an unconventional modified Chalk–Harrod type mechanism, involving the migratory insertion into the Ir–C bond and C–H reductive elimination. Both steric and electronic effects of the alkoxy group were found to account for the complete branched selectivity.
Co-reporter:Genping Huang
Organic Letters 2015 Volume 17(Issue 8) pp:1994-1997
Publication Date(Web):April 7, 2015
DOI:10.1021/acs.orglett.5b00753
The rhodium-catalyzed cyclopropanation/cyclization of allenynes was investigated by means of DFT calculations. The results show that the cyclopropanation via the proposed stepwise C(sp3)–H activation (σ-bond metathesis/C–H reductive elimination) was kinetically unfavorable. Instead, a concerted C(sp3)–H activation pathway, namely the metal-assisted σ-bond metathesis, in which the hydrogen was directly transferred to the rhodacyclopentane assisted by the Rh center followed by a C–C reductive elimination, was found to explain the experimental results.
Co-reporter:Genping Huang
The Journal of Organic Chemistry 2015 Volume 80(Issue 15) pp:7564-7571
Publication Date(Web):July 16, 2015
DOI:10.1021/acs.joc.5b01148
The rhodium-catalyzed [7 + 2] cycloaddition and cyclopropanation/cyclization of allenylcyclopentane-alkynes developed by Mukai and co-workers ( J. Am. Chem. Soc. 2012, 134, 19580−19583) represents a rare example of the metallacycle-directed selective C(sp3)-C(sp3) vs C(sp3)-H activation. In this article, the mechanism of this intriguing reaction is investigated by means of density functional theory calculations. The calculations show that the reaction is initiated by an oxidative cyclization to form the key rhodacycle intermediate. The subsequent competing β-carbon elimination/C(sp3)-C(sp2) reductive elimination and metal-assisted σ-bond metathesis/C(sp3)-C(sp3) reductive elimination lead to the [7 + 2] cycloaddition and cyclopropanation/cyclization, respectively. The calculations reproduce quite well the experimentally observed ligand-controlled selectivity, showing that both electronic and steric effects of the ligand have an important impact on the selectivity. In particular, the ligand can significantly affect energy barriers of the metal-assisted σ-bond metathesis and C–C reductive elimination but toward opposite directions, resulting in a selectivity switch between the [7 + 2] cycloaddition and cyclopropanation/cyclization upon ligand change.
Co-reporter:Mei Zhang and Genping Huang
Dalton Transactions 2016 - vol. 45(Issue 8) pp:NaN3557-3557
Publication Date(Web):2016/01/13
DOI:10.1039/C5DT04973C
The iridium-catalysed branched-selective hydroarylation of vinyl ethers represents a rare example of the branched-selective hydroarylation involving the non-styrene-type alkenes. Herein, we report our DFT calculations on the mechanism of this reaction. The results show that after C–H oxidative addition, instead of the widely accepted Chalk–Harrod type mechanism, the branched-selective hydroarylation may proceed through an unconventional modified Chalk–Harrod type mechanism, involving the migratory insertion into the Ir–C bond and C–H reductive elimination. Both steric and electronic effects of the alkoxy group were found to account for the complete branched selectivity.
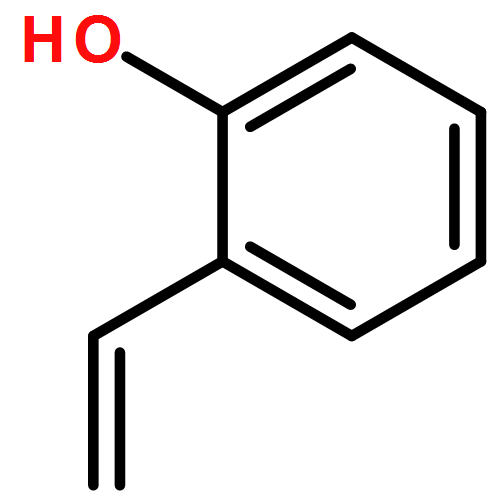
Co-reporter:Li Wang, Hongyan Zou, Xinwen Zhang and Genping Huang
Inorganic Chemistry Frontiers 2017 - vol. 4(Issue 4) pp:
Publication Date(Web):
DOI:10.1039/C6QO00671J

![<br>Bis(1,5-cyclooctadiene)iridium(I) tetrakis[3,5-bis(trifluoromethyl)phenyl]b orate](http://img.cochemist.com/ccimg/666900/666826-16-0.png)
![<br>Bis(1,5-cyclooctadiene)iridium(I) tetrakis[3,5-bis(trifluoromethyl)phenyl]b orate](http://img.cochemist.com/ccimg/666900/666826-16-0_b.png)




![Rhodium, chlorobis[1,1'-(1,2-ethanediyl)bis[1,1-diphenylphosphine-κP]]-](/data/chemimg/1735400/14874-99-8.png)
![Rhodium, chlorobis[1,1'-(1,2-ethanediyl)bis[1,1-diphenylphosphine-κP]]-](/data/chemimg/1735400/14874-99-8_b.png)
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7.png)
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7_b.png)