Co-reporter:Gaopeng Song, Xiang Zhu, Junhua Li, Dekun Hu, Dongsheng Zhao, Yixian Liao, Juntong Lin, Lian-Hui Zhang, Zi-Ning Cui
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 20(Issue 20) pp:
Publication Date(Web):15 October 2017
DOI:10.1016/j.bmc.2017.08.045
Improvement of subtype selectivity of an inhibitor’s binding activity using the conformational restriction approach has become an effective strategy in drug discovery. In this study, we applied this approach to PDE4 inhibitors and designed a series of novel oxazolidinone-fused 1,2,3,4-tetrahydroisoquinoline derivatives as conformationally restricted analogues of rolipram. The bioassay results demonstrated the oxazolidinone-fused tetrahydroisoquinoline derivatives exhibited moderate to good inhibitory activity against PDE4B and high selectivity for PDE4B/PDE4D. Among these derivatives, compound 12 showed both the strongest inhibition activity (IC50 = 0.60 μM) as well as good selectivity against PDE4B and good in vivo activity in animal models of asthma/COPD and sepsis induced by LPS. The primary SAR study showed that restricting the conformation of the catechol moiety in rolipram with the scaffold of oxazolidinone-fused tetrahydroisoquinoline could lead to an increase in selectivity for PDE4B over PDE4D, which was consistent with the observed docking simulation.Download high-res image (226KB)Download full-size image
Co-reporter:Gaopeng Song, Sumei Li, Zhiwei Lei, Yibin Li, Junhua Li, Yixian Liao and Zi-Ning Cui
Organic & Biomolecular Chemistry 2016 vol. 14(Issue 28) pp:6691-6702
Publication Date(Web):19 May 2016
DOI:10.1039/C6OB00862C
Two partially acylated oligorhamnoside derivatives 1 and 2 structurally related to the natural product mezzettiaside-6 were synthesized via a ‘2 + 1 + 1’ convergent strategy. The bioassay results showed that the introduction of the acetyl groups to the 2-position of the terminal L-rhamnose was helpful to improve in vitro cytotoxicity. Compound 1 showed both extensive in vitro cytotoxicity in tumor cell lines and potential antimultidrug resistance capability. Preliminary mechanistic studies demonstrated that compound 1 could inhibit cell growth by inducing apoptosis, arresting cell cycle progression at the S phase in K562 cells.
Co-reporter:Ya-Sheng Li, Hao Tian, Dong-Sheng Zhao, De-Kun Hu, Xing-Yu Liu, Hong-Wei Jin, Gao-Peng Song, Zi-Ning Cui
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 15) pp:3632-3635
Publication Date(Web):1 August 2016
DOI:10.1016/j.bmcl.2016.06.002
A series of pyrazole and triazole derivatives containing 5-phenyl-2-furan functionality were designed and synthesized as phosphodiesterase type 4 (PDE4) inhibitors. The bioassay results showed that title compounds exhibited considerable inhibitory activity against PDE4B and blockade of LPS-induced TNFα release. Meanwhile, the activity of compounds containing 1,2,4-triazole (series II) was higher than that of pyrazole-attached derivatives (series I). The primary structure–activity relationship study and docking results showed that the 1,2,4-triazole moiety of compound IIk played a key role to form integral hydrogen bonds and π–π stacking interaction with PDE4B protein while the rest part of the molecule extended into the catalytic domain to block the access of cAMP and formed the foundation for inhibition of PDE4. Compound IIk would be great promise as a hit compound for further study based on the preliminary structure–activity relationship and molecular modeling studies.
Co-reporter:Gao-peng Song, Su-mei Li, Hong-zong Si, Yi-bin Li, Ya-sheng Li, Ji-hong Fan, Qian-qian Liang, Hui-bing He, Han-ming Ye and Zi-ning Cui
RSC Advances 2015 vol. 5(Issue 45) pp:36092-36103
Publication Date(Web):15 Apr 2015
DOI:10.1039/C5RA02846A
A series of xanthone and thioxanthone rhamnopyranosides were designed and synthesized. Their in vitro cytotoxicity and topoisomerase inhibitory activity were evaluated. The bioassay results indicated that the introduction of the 2,3-di-O-acetyl-α-L-rhamnopyranosyl moiety to anthracene was helpful to improve the cytotoxicity in vitro. The modifications of anthracene had an important effect on the tumor cell growth inhibitory activity. Interestingly, consistency was observed between the cytotoxicity and topo I activity in these anthracene analogs, suggesting that the incorporation of either a polymethyleneamine side chain or a pyrazole ring into the anthraquinone chromophore was able to enhance topo I inhibitory activity as well as cytotoxicity simultaneously. Among them, compound 11 as a new lead compound was discovered, which showed wide in vitro cytotoxicity against 12 tumor cell lines and potential antimultidrug resistance capability. It was proved that compound 11 could induce cell apoptosis in KB cells via both extrinsic and intrinsic pathways. Flow cytometric analysis exhibited that treatment of KB cells with compound 11 led to cell apoptosis accompanied by cell cycle arrest at the G2/M phase. Furthermore, compound 11 caused a significant and dose-dependent inhibition of topoisomerase I catalytic activity.
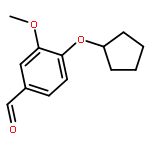
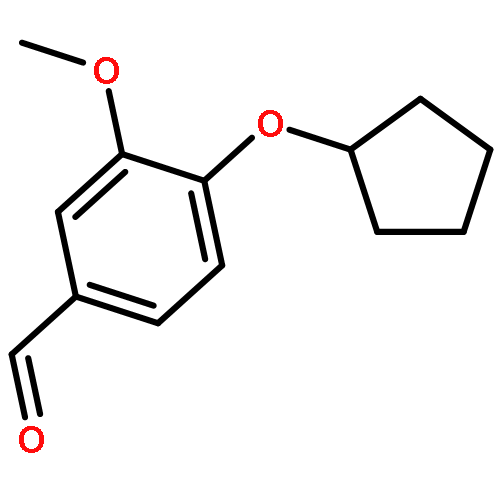
Co-reporter:Gaopeng Song, Dongsheng Zhao, Dekun Hu, Yasheng Li, Hongwei Jin, Zining Cui
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 20) pp:4610-4614
Publication Date(Web):15 October 2015
DOI:10.1016/j.bmcl.2015.08.043
The design, synthesis, and biological evaluation of new phosphodiesterase type 4 (PDE4) inhibitors, which possessed 7-(cyclopentyloxy)-6-methoxy 1,2,3,4-tetrahydroisoquinoline ring, were described. Compound 8 [(7-cyclopentyloxy)-6-methoxy-3,4-dihydroisoquinolin-2(1H)-yl)(4-hydroxy-3-methoxy-phenyl)methanone] showed the best inhibitory activity and good selectivity against PDE4B. The docking results showed that the catechol diether moiety of compound 8 played a key role to form integral hydrogen bonds with PDE4B protein while the rest part of the molecule extended into the catalytic domain to block the access of cAMP and formed the foundation for inhibition of PDE4. Compound 8 would be great promise as a hit compound for further study based on the preliminary structure–activity relationship and molecular modeling studies.
Co-reporter:Ya-Sheng Li, De-Kun Hu, Dong-Sheng Zhao, Xing-Yu Liu, Hong-Wei Jin, Gao-Peng Song, Zi-Ning Cui, Lian-Hui Zhang
Bioorganic & Medicinal Chemistry (15 March 2017) Volume 25(Issue 6) pp:
Publication Date(Web):15 March 2017
DOI:10.1016/j.bmc.2017.01.047
In this study, a series of pyrazole derivatives containing 4-phenyl-2-oxazole moiety were designed and synthesized in a concise way, some of which exhibited considerable inhibitory activity against PDE4B and blockade of LPS-induced TNF-α release. Compound 4c displayed the strongest inhibition activity (IC50 = 1.6 ± 0.4 μM) and good selectivity against PDE4B. Meanwhile, compound 4c showed good in vivo activity in animal models of asthma/COPD and sepsis induced by LPS. The primary structure–activity relationship study showed the 3,5-dimethylpyrazole residue was essential for the bioactivity, and the substituted group R1 at the benzene ring also affected the activity. Docking results showed that compound 4c played a key role to form integral hydrogen bonds and a π-π stacking interaction, using hydrazide scaffold (CONN) and pyrazole ring respectively, with PDE4B protein. While the rest part of the molecule extended into the catalytic domain to block the access of cAMP and formed the foundation for inhibition of PDE4B. Compound 4c would be great promise as a lead compound for further study based on the preliminary structure-activity relationship and molecular modeling studies.
Co-reporter:Caixiu Liu, Xiaojing Yan, Minlong Wang, Peiwen Qin, Zhiqiu Qi, Mingshan Ji, Xingyu Liu, P. Vijaya Babu, Xinghai Li, Zi-Ning Cui
Bioorganic & Medicinal Chemistry Letters (15 January 2017) Volume 27(Issue 2) pp:
Publication Date(Web):15 January 2017
DOI:10.1016/j.bmcl.2016.11.061
A series of novel 2-substituted aminocycloalkylsulfonamides were designed and synthesized by highly selective N-alkylation reaction, whose structures were characterized by 1H NMR, 13C NMR and HRMS. Among them, the configuration of compounds III12 and III20 were confirmed by X-ray single crystal diffraction. Bioassays demonstrated that the title compounds had considerable effects on different strains of Botrytis cinerea and Pyricularia grisea. Comparing with positive control procymidone (EC50 = 10.31 mg/L), compounds III28, III29, III30 and III31 showed excellent fungicidal activity against a strain of B. cinerea (CY-09), with EC50 values of 3.17, 3.04, 2.54 and 1.99 mg/L respectively. Their in vivo fungicidal activities were also better than the positive controls cyprodinil, procymidone, boscalid and carbendazim in pot experiments. Moreover, the fungicidal activity of III28 (EC50 = 4.62 mg/L) against P. grisea was also better than that of the positive control isoprothiolane (EC50 = 6.11 mg/L). Compound III28 would be great promise as a hit compound for further study based on the structure-activity relationship.
Co-reporter:Gaopeng Song, Sumei Li, Zhiwei Lei, Yibin Li, Junhua Li, Yixian Liao and Zi-Ning Cui
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 28) pp:NaN6702-6702
Publication Date(Web):2016/05/19
DOI:10.1039/C6OB00862C
Two partially acylated oligorhamnoside derivatives 1 and 2 structurally related to the natural product mezzettiaside-6 were synthesized via a ‘2 + 1 + 1’ convergent strategy. The bioassay results showed that the introduction of the acetyl groups to the 2-position of the terminal L-rhamnose was helpful to improve in vitro cytotoxicity. Compound 1 showed both extensive in vitro cytotoxicity in tumor cell lines and potential antimultidrug resistance capability. Preliminary mechanistic studies demonstrated that compound 1 could inhibit cell growth by inducing apoptosis, arresting cell cycle progression at the S phase in K562 cells.











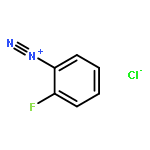





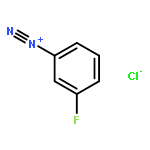
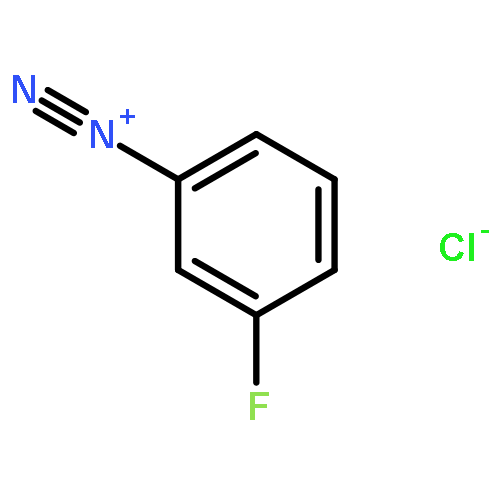
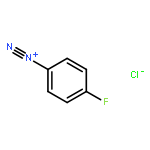












![cyano(3-phenoxyphenyl)methyl (1S,3S)-3-[(1Z)-2-chloro-3,3,3-trifluoroprop-1-en-1-yl]-2,2-dimethylcyclopropanecarboxylate](http://img.cochemist.com/ccimg/68100/68085-85-8.png)
![cyano(3-phenoxyphenyl)methyl (1S,3S)-3-[(1Z)-2-chloro-3,3,3-trifluoroprop-1-en-1-yl]-2,2-dimethylcyclopropanecarboxylate](http://img.cochemist.com/ccimg/68100/68085-85-8_b.png)

