Co-reporter:Fan-Wei Peng, Ji Xuan, Ting-Ting Wu, Jia-Yu Xue, Zi-Wei Ren, Da-Ke Liu, Xiu-Qi Wang, Xin-Hang Chen, Jia-Wei Zhang, Yun-Gen Xu, Lei Shi
European Journal of Medicinal Chemistry 2016 Volume 109() pp:1-12
Publication Date(Web):15 February 2016
DOI:10.1016/j.ejmech.2015.12.033
•24 Novel hybrids were synthesized as dual VEGFR-2/HADC inhibitors.•The in vitro enzymatic and cellular activities were evaluated.•The primary structure–activity relationships were discussed.•Compound 6fd exhibited the most potent biological activities.A single agent that simultaneously inhibits multiple targets may offer greater therapeutic benefits in cancer than single-acting agents through interference with multiple pathways and potential synergistic action. In this work, a series of hybrids bearing N-phenylquinazolin-4-amine and hydroxamic acid moieties were designed and identified as dual VEGFR-2/HDAC inhibitors. Compound 6fd exhibited the most potent inhibitory activity against HDAC with IC50 of 2.2 nM and strong inhibitory effect against VEGFR-2 with IC50 of 74 nM. It also showed the most potent inhibitory activity against a human breast cancer cell line MCF-7 with IC50 of 0.85 μM. Docking simulation supported the initial pharmacophoric hypothesis and suggested a common mode of interaction at the active binding sites of VEGFR-2 and HDLP ((Histone Deacetylase-Like Protein), which demonstrates that compound 6fd is a potential agent for cancer therapy deserving further researching.
Co-reporter:Fan-Wei Peng, Ting-Ting Wu, Zi-Wei Ren, Jia-Yu Xue, Lei Shi
Bioorganic & Medicinal Chemistry Letters 2015 25(22) pp: 5137-5141
Publication Date(Web):
DOI:10.1016/j.bmcl.2015.10.006
Co-reporter:Lei Shi, Ting-Ting Wu, Zhi Wang, Jia-Yu Xue, Yun-Gen Xu
European Journal of Medicinal Chemistry 2014 Volume 84() pp:698-707
Publication Date(Web):12 September 2014
DOI:10.1016/j.ejmech.2014.07.071
•21 Novel quinolin-4-amines containing benzimidazole moiety were synthesized.•The anticancer activities and inhibitory activities of VEGFR-2 were evaluated.•The primary structure–activity relationships were discussed.•Compound 7s exhibited the most potent inhibitory activities against VEGFR-2.•Compound 7s also showed potent anticancer activities against MCF-7 and Hep-G2.Inhibition of the VEGF signaling pathway has become a valuable approach in the treatment of cancers. In this work, a series of N-(2-phenyl-1H-benzo[d]imidazol-5-yl)quinolin-4-amine derivatives were designed and identified as potent inhibitors of VEGFR-2 (KDR) kinase. These compounds with quinoline scaffold and benzimidazole moiety were synthesized and their biological activities against VEGFR-2 and two human cancer cell lines were evaluated. Among them, compound 7s exhibited the most potent inhibitory activity against VEGFR-2 with IC50 of 0.03 μM and it also showed the highest anticancer activity against the tested cancer cell lines with IC50 of 1.2 μM against MCF-7 and 13.3 μM against Hep-G2. Docking simulation supported the initial pharmacophoric hypothesis and suggested a common mode of interaction at the ATP-binding site of VEGFR-2, which demonstrates that compound 7s is a potential agent for cancer therapy deserving further researching.A series of quinolin-4-amine derivatives containing benzimidazole moiety were discovered as potent inhibitors of VEGFR-2. Compound 7s exhibited the most potent inhibitory activity with IC50 of 0.03 μM against VEGFR-2.
Co-reporter:Lei Shi, Ting-Ting Wu, Zhi Wang, Jia-Yu Xue, Yun-Gen Xu
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 17) pp:4735-4744
Publication Date(Web):1 September 2014
DOI:10.1016/j.bmc.2014.07.008
Both c-Met and VEGFR-2 are important targets for the treatment of cancers. In this study, a series of N-(2-phenyl-1H-benzo[d]imidazol-5-yl)quinazolin-4-amine derivatives were designed and identified as dual c-Met and VEGFR-2 inhibitors. Among these compounds bearing quinazoline and benzimidazole fragments, compound 7j exhibited the most potent inhibitory activity against c-Met and VEGFR-2 with IC50 of 0.05 μM and 0.02 μM, respectively. It also showed the highest anticancer activity against the tested cancer cell lines with IC50 of 1.5 μM against MCF-7 and 8.7 μM against Hep-G2. Docking simulation supported the initial pharmacophoric hypothesis and suggested a common mode of interaction at the ATP-binding site of c-Met and VEGFR-2, which demonstrates that compound 7j is a potential agent for cancer therapy deserving further researching.A series of quinazolin-4-amine derivatives containing benzimidazole moiety were discovered as potent dual inhibitors of c-Met and VEGFR-2. Compound 7j exhibited the most potent inhibitory activities with IC50 of 0.05 μM and 0.02 μM against c-Met and VEGFR-2, respectively.



















![7,11b-dihydrobenz[b]indeno[1,2-d]pyran-3,6a,9,10(6H)-tetrol](http://img.cochemist.com/ccimg/500/474-07-7.png)
![7,11b-dihydrobenz[b]indeno[1,2-d]pyran-3,6a,9,10(6H)-tetrol](http://img.cochemist.com/ccimg/500/474-07-7_b.png)






![6-QUINAZOLINOL, 4-[(4-CHLOROPHENYL)AMINO]-7-METHOXY-](http://img.cochemist.com/ccimg/808800/808762-44-9.png)
![6-QUINAZOLINOL, 4-[(4-CHLOROPHENYL)AMINO]-7-METHOXY-](http://img.cochemist.com/ccimg/808800/808762-44-9_b.png)
![6-Quinazolinol, 4-[(3-chlorophenyl)amino]-7-methoxy-](http://img.cochemist.com/ccimg/655300/655247-78-2.png)
![6-Quinazolinol, 4-[(3-chlorophenyl)amino]-7-methoxy-](http://img.cochemist.com/ccimg/655300/655247-78-2_b.png)










![6-Quinazolinol, 4-[(3-bromophenyl)amino]-7-methoxy-](http://img.cochemist.com/ccimg/295400/295330-61-9.png)
![6-Quinazolinol, 4-[(3-bromophenyl)amino]-7-methoxy-](http://img.cochemist.com/ccimg/295400/295330-61-9_b.png)

























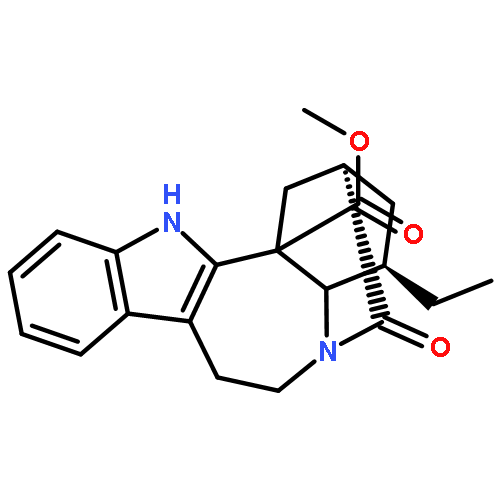
![2-Propenoic acid, 1,1'-[2-[[3-[(1-oxo-2-propen-1-yl)oxy]-2,2-bis[[(1-oxo-2-propen-1-yl)oxy]methyl]propoxy]methyl]-2-[[(1-oxo-2-propen-1-yl)oxy]methyl]-](/data/chemimg/459700/29570-58-9.png)
![2-Propenoic acid, 1,1'-[2-[[3-[(1-oxo-2-propen-1-yl)oxy]-2,2-bis[[(1-oxo-2-propen-1-yl)oxy]methyl]propoxy]methyl]-2-[[(1-oxo-2-propen-1-yl)oxy]methyl]-](/data/chemimg/459700/29570-58-9_b.png)


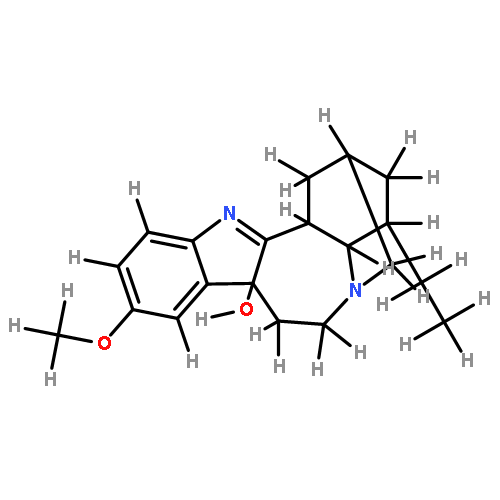
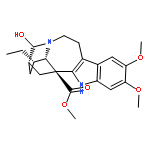
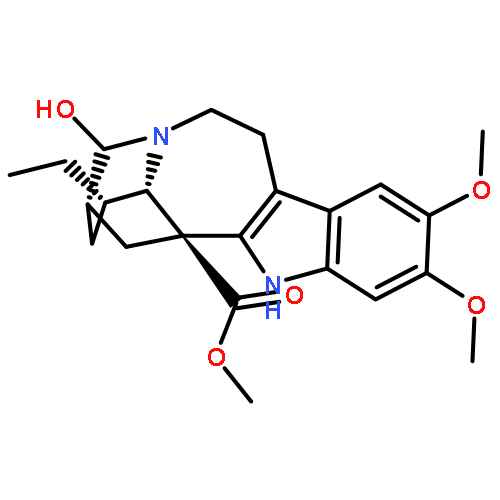
![8H-6,11b-Methanofuro[2,3-c]pyrido[1,2-a]azepin-2(6H)-one,9,10,11,11a-tetrahydro-, (6R,11aS,11bR)-](http://img.cochemist.com/ccimg/6800/6704-68-3.png)
![8H-6,11b-Methanofuro[2,3-c]pyrido[1,2-a]azepin-2(6H)-one,9,10,11,11a-tetrahydro-, (6R,11aS,11bR)-](http://img.cochemist.com/ccimg/6800/6704-68-3_b.png)






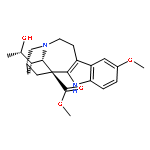









![2-(4-Bromophenyl)-1H-benzo[d]imidazol-5-amine](http://img.cochemist.com/ccimg/351300/351226-76-1.png)
![2-(4-Bromophenyl)-1H-benzo[d]imidazol-5-amine](http://img.cochemist.com/ccimg/351300/351226-76-1_b.png)
![1H-Benzimidazol-5-amine, 2-[3-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/263100/263022-20-4.png)
![1H-Benzimidazol-5-amine, 2-[3-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/263100/263022-20-4_b.png)
![1H-Benzimidazol-5-amine, 2-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/263100/263022-04-4.png)
![1H-Benzimidazol-5-amine, 2-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/263100/263022-04-4_b.png)




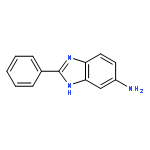
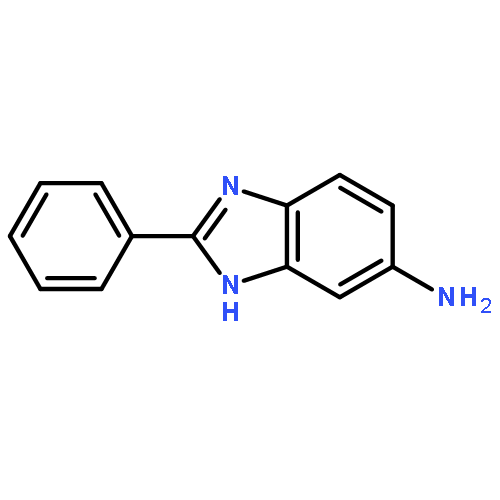
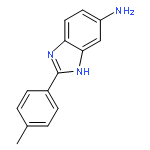
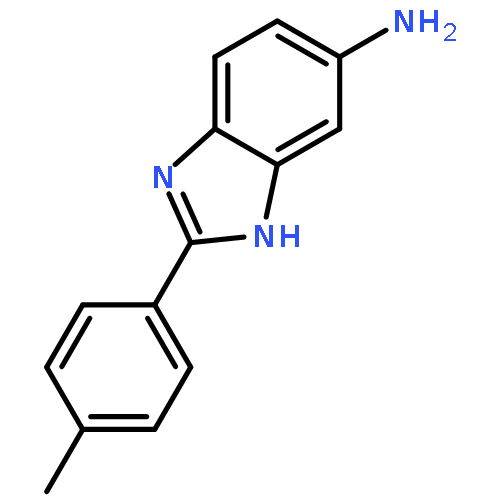
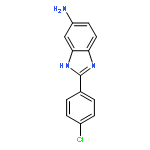
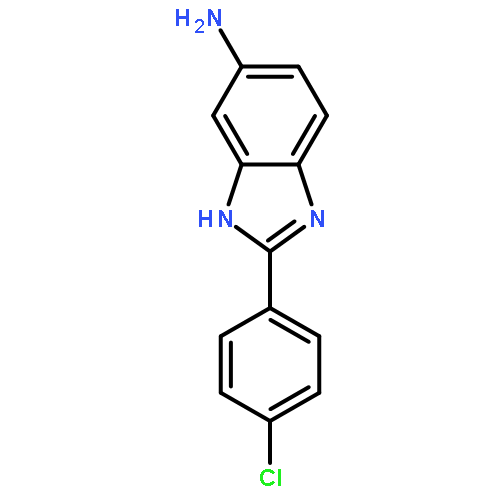
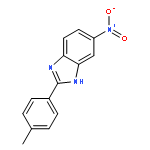
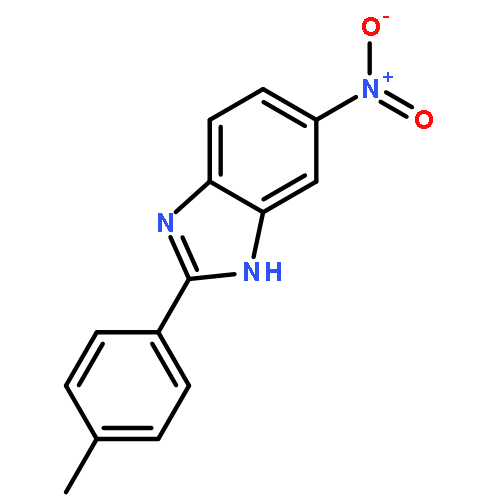
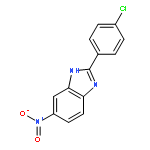
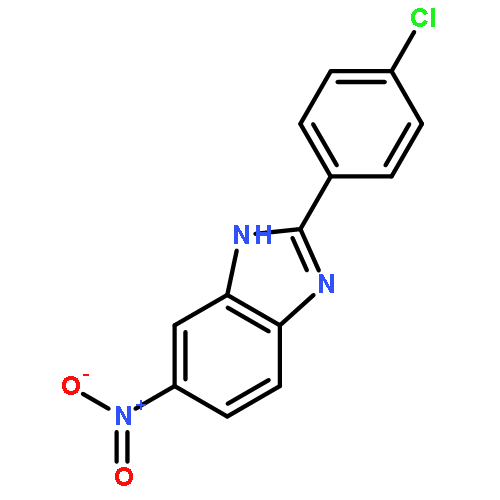
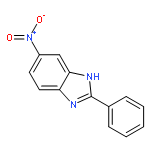
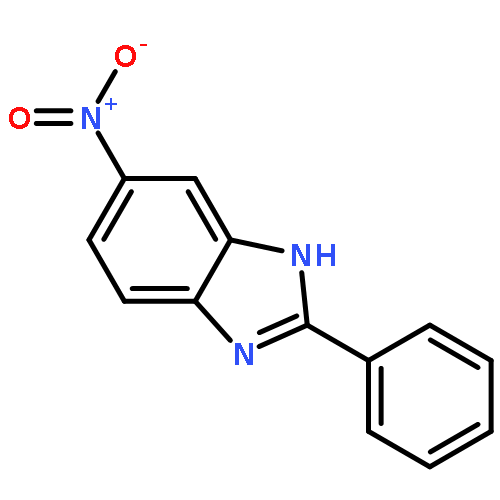
![3-{4-[1-(4-FLUOROPHENYL)-2,5-DIOXO-3-PYRROLIDINYL]-1-PIPERAZINYL}<WBR />PROPANENITRILE](http://img.cochemist.com/ccimg/400/348-35-6.png)
![3-{4-[1-(4-FLUOROPHENYL)-2,5-DIOXO-3-PYRROLIDINYL]-1-PIPERAZINYL}<WBR />PROPANENITRILE](http://img.cochemist.com/ccimg/400/348-35-6_b.png)
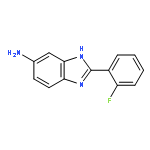
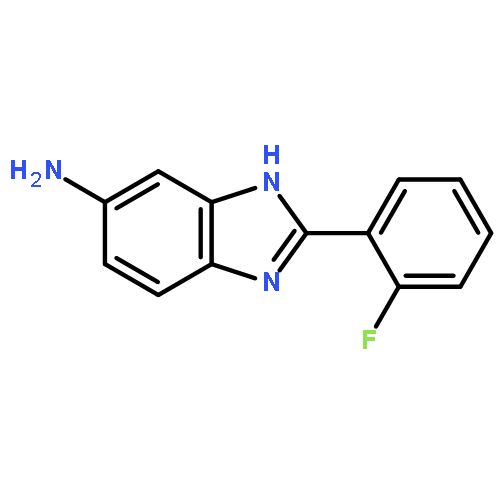
![2-(2-fluorophenyl)-5-nitro-1H-benzo[d]imidazole](http://img.cochemist.com/ccimg/400/348-33-4.png)
![2-(2-fluorophenyl)-5-nitro-1H-benzo[d]imidazole](http://img.cochemist.com/ccimg/400/348-33-4_b.png)