Co-reporter:Nilkamal Mahanta, Zhengan Zhang, Graham A. Hudson, Wilfred A. van der Donk, and Douglas A. Mitchell
Journal of the American Chemical Society March 29, 2017 Volume 139(Issue 12) pp:4310-4310
Publication Date(Web):March 16, 2017
DOI:10.1021/jacs.7b00693
Thiomuracin is a thiopeptide antibiotic with potent activity toward Gram-positive drug-resistant bacteria. Thiomuracin is biosynthesized from a precursor peptide, TbtA, by a complex array of posttranslational modifications. One of several intriguing transformations is the C-methylation of thiazole, occurring at an unactivated sp2 carbon. Herein, we report the in vitro reconstitution of TbtI, the responsible radical S-adenosyl-methionine (rSAM) C-methyltransferase, which catalyzes the formation of 5-methylthiazole at a single site. Our studies demonstrate that a linear hexazole-bearing intermediate of TbtA is a substrate for TbtI whereas macrocyclized thiomuracin GZ is not. In determining the minimal substrate for TbtI, we found that the enzyme is functional when most of the leader peptide has been removed. The in vitro reconstitution of TbtI, a class C rSAM methyltransferase, further adds to the chemical versatility of rSAM enzymes, and informs on the complexity of thiomuracin biosynthesis.
Co-reporter:Brandon J. Burkhart, Nidhi Kakkar, Graham A. Hudson, Wilfred A. van der Donk, and Douglas A. Mitchell
ACS Central Science June 28, 2017 Volume 3(Issue 6) pp:629-629
Publication Date(Web):June 6, 2017
DOI:10.1021/acscentsci.7b00141
Combining biosynthetic enzymes from multiple pathways is an attractive approach for producing molecules with desired structural features; however, progress has been hampered by the incompatibility of enzymes from unrelated pathways and intolerance toward alternative substrates. Ribosomally synthesized and posttranslationally modified peptides (RiPPs) are a diverse natural product class that employs a biosynthetic logic that is highly amenable to engineering new compounds. RiPP biosynthetic proteins modify their substrates by binding to a motif typically located in the N-terminal leader region of the precursor peptide. Here, we exploit this feature by designing leader peptides that enable recognition and processing by multiple enzymes from unrelated RiPP pathways. Using this broadly applicable strategy, a thiazoline-forming cyclodehydratase was combined with enzymes from the sactipeptide and lanthipeptide families to create new-to-nature hybrid RiPPs. We also provide insight into design features that enable control over the hybrid biosynthesis to optimize enzyme compatibility and establish a general platform for engineering additional hybrid RiPPs.
Co-reporter:Brandon J. Burkhart, Christopher J. Schwalen, Greg Mann, James H. Naismith, and Douglas A. Mitchell
Chemical Reviews April 26, 2017 Volume 117(Issue 8) pp:5389-5389
Publication Date(Web):March 3, 2017
DOI:10.1021/acs.chemrev.6b00623
With advances in sequencing technology, uncharacterized proteins and domains of unknown function (DUFs) are rapidly accumulating in sequence databases and offer an opportunity to discover new protein chemistry and reaction mechanisms. The focus of this review, the formerly enigmatic YcaO superfamily (DUF181), has been found to catalyze a unique phosphorylation of a ribosomal peptide backbone amide upon attack by different nucleophiles. Established nucleophiles are the side chains of Cys, Ser, and Thr which gives rise to azoline/azole biosynthesis in ribosomally synthesized and posttranslationally modified peptide (RiPP) natural products. However, much remains unknown about the potential for YcaO proteins to collaborate with other nucleophiles. Recent work suggests potential in forming thioamides, macroamidines, and possibly additional post-translational modifications. This review covers all knowledge through mid-2016 regarding the biosynthetic gene clusters (BGCs), natural products, functions, mechanisms, and applications of YcaO proteins and outlines likely future research directions for this protein superfamily.
Co-reporter:Zhengan Zhang, Graham A. Hudson, Nilkamal Mahanta, Jonathan I. Tietz, Wilfred A. van der Donk, and Douglas A. Mitchell
Journal of the American Chemical Society 2016 Volume 138(Issue 48) pp:15511-15514
Publication Date(Web):October 4, 2016
DOI:10.1021/jacs.6b08987
The biosynthesis of the thiopeptide thiomuracin is a well-orchestrated process involving a multitude of posttranslational modifications. We show that six Cys residues of a precursor peptide are first cyclodehydrated and oxidized to thiazoles in an ordered, but nonlinear fashion that is leader-peptide-dependent. Then four alcohols are glutamylated and converted to alkenes in a C-to-N terminal directional process that is leader-peptide-independent. Finally, two of these alkenes undergo a formal [4 + 2] cycloaddition to form a trithiazole-substituted pyridine macrocycle. We describe here the factors that govern the substrate specificity and order of biosynthetic events that turn a ribosomal peptide into a powerful antibiotic.
Co-reporter:Tucker Maxson, Jonathan I. Tietz, Graham A. Hudson, Xiao Rui Guo, Hua-Chia Tai, and Douglas A. Mitchell
Journal of the American Chemical Society 2016 Volume 138(Issue 46) pp:15157-15166
Publication Date(Web):October 31, 2016
DOI:10.1021/jacs.6b06848
Natural products (NPs) serve important roles as drug candidates and as tools for chemical biology. However, traditional NP discovery, largely based on bioassay-guided approaches, is biased toward abundant compounds and rediscovery rates are high. Orthogonal methods to facilitate discovery of new NPs are thus needed, and herein we describe an isotope tag-based expansion of reactivity-based NP screening to address these shortcomings. Reactivity-based screening is a directed discovery approach in which a specific reactive handle on the NP is targeted by a chemoselective probe to enable its detection by mass spectrometry. In this study, we have developed an aminooxy-containing probe to guide the discovery of aldehyde- and ketone-containing NPs. To facilitate the detection of labeling events, the probe was dibrominated, imparting a unique isotopic signature to distinguish labeled metabolites from spectral noise. As a proof of concept, the probe was then utilized to screen a collection of bacterial extracts, leading to the identification of a new analogue of antipain, deimino-antipain. The bacterial producer of deimino-antipain was sequenced and the responsible biosynthetic gene cluster was identified by bioinformatic analysis and heterologous expression. These data reveal the previously undetermined genetic basis for a well-known family of aldehyde-containing, peptidic protease inhibitors, including antipain, chymostatin, leupeptin, elastatinal, and microbial alkaline protease inhibitor, which have been widely used for over 40 years.
Co-reporter:Caitlin D. Deane, Brandon J. Burkhart, Patricia M. Blair, Jonathan I. Tietz, Alice Lin, and Douglas A. Mitchell
ACS Chemical Biology 2016 Volume 11(Issue 8) pp:2232
Publication Date(Web):June 1, 2016
DOI:10.1021/acschembio.6b00369
Plantazolicin (PZN) is a ribosomally synthesized and post-translationally modified peptide (RiPP) natural product that exhibits extraordinarily narrow-spectrum antibacterial activity toward the causative agent of anthrax, Bacillus anthracis. During PZN biosynthesis, a cyclodehydratase catalyzes cyclization of cysteine, serine, and threonine residues in the PZN precursor peptide (BamA) to azolines. Subsequently, a dehydrogenase oxidizes most of these azolines to thiazoles and (methyl)oxazoles. The final biosynthetic steps consist of leader peptide removal and dimethylation of the nascent N-terminus. Using a heterologously expressed and purified heterocycle synthetase, the BamA peptide was processed in vitro concordant with the pattern of post-translational modification found in the naturally occurring compound. Using a suite of BamA-derived peptides, including amino acid substitutions as well as contracted and expanded substrate variants, the substrate tolerance of the heterocycle synthetase was elucidated in vitro, and the residues crucial for leader peptide binding were identified. Despite increased promiscuity compared to what was previously observed during heterologous production in E. coli, the synthetase retained exquisite selectivity in cyclization of unnatural peptides only at positions which correspond to those cyclized in the natural product. A cleavage site was subsequently introduced to facilitate leader peptide removal, yielding mature PZN variants after enzymatic or chemical dimethylation. In addition, we report the isolation and characterization of two novel PZN-like natural products that were predicted from genome sequences but whose production had not yet been observed.
Co-reporter:Katie J. Molohon, Patricia M. Blair, Seongjin Park, James R. Doroghazi, Tucker Maxson, Jeremy R. Hershfield, Kristen M. Flatt, Nathan E. Schroeder, Taekjip Ha, and Douglas A. Mitchell
ACS Infectious Diseases 2016 Volume 2(Issue 3) pp:207
Publication Date(Web):December 23, 2015
DOI:10.1021/acsinfecdis.5b00115
Plantazolicin (PZN) is a ribosomally synthesized and post-translationally modified natural product from Bacillus methylotrophicus FZB42 and Bacillus pumilus. Extensive tailoring to 12 of the 14 amino acid residues in the mature natural product endows PZN with not only a rigid, polyheterocyclic structure, but also antibacterial activity. Here we report the remarkably discriminatory activity of PZN toward Bacillus anthracis, which rivals a previously described gamma (γ) phage lysis assay in distinguishing B. anthracis from other members of the Bacillus cereus group. We evaluate the underlying cause of this selective activity by measuring the RNA expression profile of PZN-treated B. anthracis, which revealed significant up-regulation of genes within the cell envelope stress response. PZN depolarizes the B. anthracis membrane like other cell envelope-acting compounds but uniquely localizes to distinct foci within the envelope. Selection and whole-genome sequencing of PZN-resistant mutants of B. anthracis implicate a relationship between the action of PZN and cardiolipin (CL) within the membrane. Exogenous CL increases the potency of PZN in wild type B. anthracis and promotes the incorporation of fluorescently tagged PZN in the cell envelope. We propose that PZN localizes to and exacerbates structurally compromised regions of the bacterial membrane, which ultimately results in cell lysis.Keywords: anthrax; antibiotic; Bacillus anthracis; membrane depolarization; mode of action; oxazole; pathogen specific antibiotic; ribosomally synthesized and post-translationally modified natural product; thiazole
Co-reporter:Tucker Maxson, Douglas A. Mitchell
Tetrahedron 2016 Volume 72(Issue 25) pp:3609-3624
Publication Date(Web):23 June 2016
DOI:10.1016/j.tet.2015.09.069
Co-reporter:Kyle L. Dunbar; Jonathan I. Tietz; Courtney L. Cox; Brandon J. Burkhart
Journal of the American Chemical Society 2015 Volume 137(Issue 24) pp:7672-7677
Publication Date(Web):May 29, 2015
DOI:10.1021/jacs.5b04682
Thiazole/oxazole-modified microcins (TOMMs) are a class of post-translationally modified peptide natural products bearing azole and azoline heterocycles. The first step in heterocycle formation is carried out by a two component cyclodehydratase comprised of an E1 ubiquitin-activating and a YcaO superfamily member. Recent studies have demonstrated that the YcaO domain is responsible for cyclodehydration, while the TOMM E1 homologue is responsible for peptide recognition during azoline formation. Although all characterized TOMM biosynthetic clusters contain this canonical TOMM E1 homologue (C domain), we also identified a second, highly divergent E1 superfamily member, annotated as an Ocin-ThiF-like protein (F protein), associated with more than 300 TOMM biosynthetic clusters. Here we describe the in vitro reconstitution of a novel TOMM cyclodehydratase from such a cluster and demonstrate that this auxiliary protein is required for cyclodehydration. Using a combination of biophysical techniques, we demonstrate that the F protein, rather than the C domain, is responsible for engaging the peptide substrate. The C domain instead appears to serve as a scaffolding protein, bringing the catalytic YcaO domain and the peptide binding Ocin-ThiF-like protein into proximity. Our findings provide an updated biosynthetic framework that provides a foundation for the characterization and reconstitution of approximately 25% of bioinformatically identifiable TOMM synthetases.
Co-reporter:Graham A. Hudson; Zhengan Zhang; Jonathan I. Tietz; Douglas A. Mitchell;Wilfred A. van der Donk
Journal of the American Chemical Society 2015 Volume 137(Issue 51) pp:16012-16015
Publication Date(Web):December 17, 2015
DOI:10.1021/jacs.5b10194
Thiopeptides are potent antibiotics that inhibit protein synthesis. They are made by a remarkable post-translational modification process that transforms a linear peptide into a polycyclic structure. We present here the in vitro biosynthesis of the core scaffold of thiomuracin catalyzed by six proteins. We show that cyclodehydration precedes dehydration, and that dehydration is catalyzed by two proteins in a tRNAGlu-dependent manner. The enzyme that generates the pyridine core from two dehydroalanines ejects the leader peptide as a C-terminal carboxamide. Mutagenesis studies of the enzyme TbtD identified important residues for a formal [4+2] cycloaddition process. The core structure of thiomuracin exhibits similar antimicrobial activity to other known congeners, illustrating that in vitro biosynthesis is a viable route to potent antibiotics that can be explored for the rapid and renewable generation of analogues.
Co-reporter:Mikhail Metelev, Jonathan I. Tietz, Joel O. Melby, Patricia M. Blair, Lingyang Zhu, Itamar Livnat, Konstantin Severinov, Douglas A. Mitchell
Chemistry & Biology 2015 Volume 22(Issue 2) pp:241-250
Publication Date(Web):19 February 2015
DOI:10.1016/j.chembiol.2014.11.017
•Streptomonomicin (STM) is a lasso peptide with a unique Ser1-Asp9 linkage•STM exhibits activity against Gram-positive bacteria•Substitutions in the essential two-component regulator WalR endow STM resistance•Genome sequencing shows biosynthetic potential in the genus StreptomonosporaNatural products are the most historically significant source of compounds for drug development. However, unacceptably high rates of compound rediscovery associated with large-scale screening of common microbial producers have resulted in the abandonment of many natural product drug discovery efforts, despite the increasing prevalence of clinically problematic antibiotic resistance. Screening of underexplored taxa represents one strategy to avoid rediscovery. Herein we report the discovery, isolation, and structural elucidation of streptomonomicin (STM), an antibiotic lasso peptide from Streptomonospora alba, and report the genome for its producing organism. STM-resistant clones of Bacillus anthracis harbor mutations to walR, the gene encoding a response regulator for the only known widely distributed and essential two-component signal transduction system in Firmicutes. To the best of our knowledge, Streptomonospora had been hitherto biosynthetically and genetically uncharacterized, with STM being the first reported compound from the genus. Our results demonstrate that understudied microbes remain fruitful reservoirs for the rapid discovery of novel, bioactive natural products.Figure optionsDownload full-size imageDownload high-quality image (179 K)Download as PowerPoint slide
Co-reporter:Yue Hao, Patricia M. Blair, Abhishek Sharma, Douglas A. Mitchell, and Satish K. Nair
ACS Chemical Biology 2015 Volume 10(Issue 5) pp:1209
Publication Date(Web):January 30, 2015
DOI:10.1021/cb501042a
Peptide antibiotics represent a class of conformationally constrained natural products of growing pharmaceutical interest. Plantazolicin (PZN) is a linear, polyheterocyclic natural product with highly selective and potent activity against the anthrax-causing bacterium, Bacillus anthracis. The bioactivity of PZN is contingent on dimethylation of its N-terminal Arg residue by an S-adenosylmethionine-dependent methyltransferase. Here, we explore the substrate tolerances of two homologous PZN methyltransferases by carrying out kinetic analyses of the enzymes against a synthetic panel of truncated PZN analogs containing the N-terminal Arg residue. X-ray cocrystal structures of the PZN methyltransferases with each of these heterocycle-containing substrates provide a rationale for understanding the strict substrate specificity of these enzymes. Kinetic studies of structure-guided, site-specific variants allowed for the assignment of residues governing catalysis and substrate scope. Microbiological testing further revealed that upon dimethylation of the N-terminal Arg, a pentaheterocyclized PZN analog retained potent anti-B. anthracis activity, nearly equal to that of full-length PZN. These studies may be useful in the biosynthetic engineering of natural product analogs with different bioactivity profiles, as demonstrated by our identification of a truncated plantazolicin derivative that is active against methicillin-resistant Staphylococcus aureus (MRSA).
Co-reporter:Tucker Maxson, Caitlin D. Deane, Evelyn M. Molloy, Courtney L. Cox, Andrew L. Markley, Shaun W. Lee, and Douglas A. Mitchell
ACS Chemical Biology 2015 Volume 10(Issue 5) pp:1217
Publication Date(Web):February 10, 2015
DOI:10.1021/cb500843r
Streptolysin S (SLS) is a post-translationally modified peptide cytolysin that is produced by the human pathogen Streptococcus pyogenes. SLS belongs to a large family of azole-containing natural products that are biosynthesized via an evolutionarily conserved pathway. SLS is an important virulence factor during S. pyogenes infections, but despite an extensive history of study, further investigations are needed to clarify several steps of its biosynthesis. To this end, chemical inhibitors of SLS biosynthesis would be valuable tools to interrogate the various maturation steps of both SLS and biosynthetically related natural products. Such chemical inhibitors could also potentially serve as antivirulence therapeutics, which in theory may alleviate the spread of antibiotic resistance. In this work, we demonstrate that FDA-approved HIV protease inhibitors, especially nelfinavir, block a key proteolytic processing step during SLS production. This inhibition was demonstrated in live S. pyogenes cells and through in vitro protease inhibition assays. A panel of 57 nelfinavir analogs was synthesized, leading to a series of compounds with improved anti-SLS activity while illuminating structure–activity relationships. Nelfinavir was also found to inhibit the maturation of other azole-containing natural products, namely those involved in listeriolysin S, clostridiolysin S, and plantazolicin production. The use of nelfinavir analogs as inhibitors of SLS production has allowed us to begin examining the proteolysis event in SLS maturation and will aid in further investigations of the biosynthesis of SLS and related natural products.
Co-reporter:Courtney L. Cox, Jonathan I. Tietz, Karol Sokolowski, Joel O. Melby, James R. Doroghazi, and Douglas A. Mitchell
ACS Chemical Biology 2014 Volume 9(Issue 9) pp:2014
Publication Date(Web):June 17, 2014
DOI:10.1021/cb500324n
Natural products remain an important source of drug candidates, but the difficulties inherent to traditional isolation, coupled with unacceptably high rates of compound rediscovery, limit the pace of natural product detection. Here we describe a reactivity-based screening method to rapidly identify exported bacterial metabolites that contain dehydrated amino acids (i.e., carbonyl- or imine-activated alkenes), a common motif in several classes of natural products. Our strategy entails the use of a commercially available thiol, dithiothreitol, for the covalent labeling of activated alkenes by nucleophilic 1,4-addition. Modification is easily discerned by comparing mass spectra of reacted and unreacted cell surface extracts. When combined with bioinformatic analysis of putative natural product gene clusters, targeted screening and isolation can be performed on a prioritized list of strains. Moreover, known compounds are easily dereplicated, effectively eliminating superfluous isolation and characterization. As a proof of principle, this labeling method was used to identify known natural products belonging to the thiopeptide, lanthipeptide, and linaridin classes. Further, upon screening a panel of only 23 actinomycetes, we discovered and characterized a novel thiopeptide antibiotic, cyclothiazomycin C.
Co-reporter:Joel O. Melby, Xiangpo Li, and Douglas A. Mitchell
Biochemistry 2014 Volume 53(Issue 2) pp:
Publication Date(Web):December 23, 2013
DOI:10.1021/bi401529y
Thiazole/oxazole-modified microcins (TOMMs) comprise a structurally diverse family of natural products with varied bioactivities linked by the presence of posttranslationally installed thiazol(in)e and oxazol(in)e heterocycles. The detailed investigation of the TOMM biosynthetic enzymes from Bacillus sp. Al Hakam (Balh) has provided significant insight into heterocycle biosynthesis. Thiazoles and oxazoles are installed by the successive action of an ATP-dependent cyclodehydratase (C- and D-protein) and a FMN-dependent dehydrogenase (B-protein), which are responsible for azoline formation and azoline oxidation, respectively. Although several studies have focused on the mechanism of azoline formation, many details regarding the role of the dehydrogenase (B-protein) in overall substrate processing remain unknown. In this work, we evaluated the involvement of the dehydrogenase in determining the order of ring formation as well as the promiscuity of the Balh and microcin B17 cyclodehydratases to accept a panel of noncognate dehydrogenases. In support of the observed promiscuity, a fluorescence polarization assay was utilized to measure binding of the dehydrogenase to the cyclodehydratase using the intrinsic fluorescence of the FMN cofactor. Ultimately, the noncognate dehydrogenases were shown to possess cyclodehydratase-independent activity. A previous study identified a conserved Lys–Tyr motif to be important for dehydrogenase activity. Using the tools developed in this study, the Lys–Tyr motif was shown neither to alter complex formation with the cyclodehydratase nor the reduction potential. Taken together with the known crystal structure of a homologue, our data suggest that the Lys–Tyr motif is of catalytic importance. Overall, this study provides a greater level of insight into the complex orchestration of enzymatic activity during TOMM biosynthesis.
Co-reporter:Caitlin D. Deane
Journal of Industrial Microbiology & Biotechnology 2014 Volume 41( Issue 2) pp:315-331
Publication Date(Web):2014 February
DOI:10.1007/s10295-013-1361-8
Natural product discovery is currently undergoing a transformation from a phenotype-driven field to a genotype-driven one. The increasing availability of genome sequences, coupled with improved techniques for identifying biosynthetic gene clusters, has revealed that secondary metabolomes are strikingly vaster than previously thought. New approaches to correlate biosynthetic gene clusters with the compounds they produce have facilitated the production and isolation of a rapidly growing collection of what we refer to as “reverse-discovered” natural products, in analogy to reverse genetics. In this review, we present an extensive list of reverse-discovered natural products and discuss seven important lessons for natural product discovery by genome-guided methods: structure prediction, accurate annotation, continued study of model organisms, avoiding genome-size bias, genetic manipulation, heterologous expression, and potential engineering of natural product analogs.
Co-reporter:Kyle L. Dunbar
Journal of the American Chemical Society 2013 Volume 135(Issue 23) pp:8692-8701
Publication Date(Web):May 20, 2013
DOI:10.1021/ja4029507
Current strategies for generating peptides and proteins bearing amide carbonyl derivatives rely on solid-phase peptide synthesis for amide functionalization. Although such strategies have been successfully implemented, technical limitations restrict both the length and sequence of the synthetic fragments. Herein we report the repurposing of a thiazole/oxazole-modified microcin (TOMM) cyclodehydratase to site-specifically install amide backbone labels onto diverse peptide substrates, a method we refer to as azoline-mediated peptide backbone labeling (AMPL). This convenient chemoenzymatic strategy can generate both thioamides and amides with isotopically labeled oxygen atoms. Moreover, we demonstrate the first leader peptide-independent activity of a TOMM synthetase, circumventing the requirement that sequences of interest be fused to a leader peptide for modification. Through bioinformatics-guided site-directed mutagenesis, we also convert a strictly dehydrogenase-dependent TOMM azole synthetase into an azoline synthetase. This vastly expands the spectrum of substrates modifiable by AMPL by allowing any in vitro reconstituted TOMM synthetase to be employed. To demonstrate the utility of AMPL for mechanistic enzymology studies, an 18O-labeled substrate was generated to provide direct evidence that cyclodehydrations in TOMMs occur through the phosphorylation of the carbonyl oxygen preceding the cyclized residue. Furthermore, we demonstrate that AMPL is a useful tool for establishing the location of azolines both on in vitro modified peptides and azoline-containing natural products.
Co-reporter:Abhishek Sharma, Patricia M. Blair, and Douglas A. Mitchell
Organic Letters 2013 Volume 15(Issue 19) pp:5076-5079
Publication Date(Web):September 24, 2013
DOI:10.1021/ol402444a
A convergent strategy for the synthesis of truncated analogues of plantazolicin (PZN), a member of the thiazole/oxazole-modified microcin (TOMM) class of natural products, has been developed. These N-terminal mono-, tri-, and pentazole substructures of PZN were utilized to probe the substrate requirements and thermodynamic ligand binding parameters of an unusually selective PZN methyltransferase (BamL) by isothermal titration calorimetry. Our results demonstrate that the presence of a single N-terminal azole permits efficient processing by BamL; however, the substrate binding becomes stronger with increased polyazole chain length.
Co-reporter:Caitlin D. Deane, Joel O. Melby, Katie J. Molohon, Aziz R. Susarrey, and Douglas A. Mitchell
ACS Chemical Biology 2013 Volume 8(Issue 9) pp:1998
Publication Date(Web):June 24, 2013
DOI:10.1021/cb4003392
Plantazolicin (PZN) is a polyheterocyclic natural product derived from a ribosomal peptide that harbors remarkable antibiotic selectivity for the causative agent of anthrax, Bacillus anthracis. To simultaneously establish the structure–activity relationship of PZN and the substrate tolerance of the biosynthetic pathway, an Escherichia coli expression strain was engineered to heterologously produce PZN analogues. Variant PZN precursor genes were produced by site-directed mutagenesis and later screened by mass spectrometry to assess post-translational modification and export by E. coli. From a screen of 72 precursor peptides, 29 PZN variants were detected. This analogue collection provided insight into the selectivity of the post-translational modifying enzymes and established the boundaries of the natural biosynthetic pathway. Unlike other studied thiazole/oxazole-modified microcins, the biosynthetic machinery appeared to be finely tuned toward the production of PZN, such that the cognate enzymes did not process even other naturally occurring sequences from similar biosynthetic clusters. The modifying enzymes were exquisitely selective, installing heterocycles only at predefined positions within the precursor peptides while leaving neighboring residues unmodified. Nearly all substitutions at positions normally harboring heterocycles prevented maturation of a PZN variant, though some exceptions were successfully produced lacking a heterocycle at the penultimate residue. No variants containing additional heterocycles were detected, although several peptide sequences yielded multiple PZN variants as a result of varying oxidation states of select residues. Eleven PZN variants were produced in sufficient quantity to facilitate purification and assessment of their antibacterial activity, providing insight into the structure–activity relationship of PZN.
Co-reporter:Kyle L. Dunbar and Douglas A. Mitchell
ACS Chemical Biology 2013 Volume 8(Issue 3) pp:473
Publication Date(Web):January 3, 2013
DOI:10.1021/cb3005325
Ribosomally synthesized posttranslationally modified peptides (RiPPs) are a rapidly growing class of natural products with diverse structures and activities. In recent years, a great deal of progress has been made in elucidating the biosynthesis of various RiPP family members. As with the study of nonribosomal peptide and polyketide biosynthetic enzymes, these investigations have led to the discovery of entirely new biological chemistry. With each unique enzyme investigated, a more complex picture of Nature’s synthetic potential is revealed. This Review focuses on recent reports (since 2008) that have changed the way that we think about ribosomal natural product biosynthesis and the enzymology of complex bond-forming reactions.
Co-reporter:Jaeheon Lee;Brandon J. Burkhart;Patricia M. Blair;Yue Hao;Vinayak Agarwal;Joel O. Melby;Satish K. Nair
PNAS 2013 Volume 110 (Issue 32 ) pp:12954-12959
Publication Date(Web):2013-08-06
DOI:10.1073/pnas.1306101110
Plantazolicin (PZN), a polyheterocyclic, Nα,Nα-dimethylarginine–containing antibiotic, harbors remarkably specific bactericidal activity toward strains of Bacillus anthracis, the causative agent of anthrax. Previous studies demonstrated that genetic deletion of the S-adenosyl-l-methionine–dependent methyltransferase from the PZN biosynthetic gene cluster results in the formation of desmethylPZN, which
is devoid of antibiotic activity. Here we describe the in vitro reconstitution, mutational analysis, and X-ray crystallographic
structure of the PZN methyltransferase. Unlike all other known small molecule methyltransferases, which act upon diverse substrates
in vitro, the PZN methyltransferase is uncharacteristically limited in substrate scope and functions only on desmethylPZN
and close derivatives. The crystal structures of two related PZN methyltransferases, solved to 1.75 Å (Bacillus amyloliquefaciens) and 2.0 Å (Bacillus pumilus), reveal a deep, narrow cavity, putatively functioning as the binding site for desmethylPZN. The narrowness of this cavity
provides a framework for understanding the molecular basis of the extreme substrate selectivity. Analysis of a panel of point
mutations to the methyltransferase from B. amyloliquefaciens allowed the identification of residues of structural and catalytic importance. These findings further our understanding of
one set of orthologous enzymes involved in thiazole/oxazole-modified microcin biosynthesis, a rapidly growing sector of natural
products research.
Co-reporter:Joel O. Melby ; Kyle L. Dunbar ; Nhat Q. Trinh
Journal of the American Chemical Society 2012 Volume 134(Issue 11) pp:5309-5316
Publication Date(Web):March 8, 2012
DOI:10.1021/ja211675n
The thiazole/oxazole-modified microcins (TOMMs) represent a burgeoning class of ribosomal natural products decorated with thiazoles and (methyl)oxazoles originating from cysteines, serines, and threonines. The ribosomal nature of TOMMs allows for the generation of derivative products from mutations in the amino acid sequence of the precursor peptide, which ultimately manifest in differing structures and, sometimes, biological functions. Employing a TOMM system for the purpose of creating new structures and functions via combinatorial biosynthesis requires processing machinery that can tolerate highly variable substrates. In this study, TOMM enzymatic promiscuity was assessed using a currently uncharacterized cluster in Bacillus sp. Al Hakam. As determined by Fourier transform tandem mass spectrometry (FT-MS/MS), azole rings were formed in both a regio- and chemoselective fashion. Cognate and noncognate precursor peptides were modified in an overall C- to N-terminal directionality, which to date is unique among characterized ribosomal natural products. Studies focused on the inherent promiscuity of the biosynthetic machinery elucidated a modest bias for glycine at the preceding (−1) position and a remarkable flexibility in the following (+1) position, even allowing for the incorporation of charged amino acids and bisheterocyclization. Two unnatural substrates were utilized as the conclusive test of substrate flexibility, of which both were processed in a predictable fashion. A greater understanding of substrate processing and enzymatic tolerance toward unnatural substrates will prove beneficial when designing combinatorial libraries to screen for artificial TOMMs that exhibit desired activities.
Co-reporter:Joel O Melby, Nathan J Nard, Douglas A Mitchell
Current Opinion in Chemical Biology 2011 Volume 15(Issue 3) pp:369-378
Publication Date(Web):June 2011
DOI:10.1016/j.cbpa.2011.02.027
With billions of years of evolution under its belt, Nature has been expanding and optimizing its biosynthetic capabilities. Chemically complex secondary metabolites continue to challenge and inspire today's most talented synthetic chemists. A brief glance at these natural products, especially the substantial structural variation within a class of compounds, clearly demonstrates that Nature has long played the role of medicinal chemist. The recent explosion in genome sequencing has expanded our appreciation of natural product space and the vastness of uncharted territory that remains. One small corner of natural product chemical space is occupied by the recently dubbed thiazole/oxazole-modified microcins (TOMMs), which are ribosomally produced peptides with posttranslationally installed heterocycles derived from cysteine, serine and threonine residues. As with other classes of natural products, the genetic capacity to synthesize TOMMs has been widely disseminated among bacteria. Over the evolutionary timescale, Nature has tested countless random mutations and selected for gain of function in TOMM biosynthetic gene clusters, yielding several privileged molecular scaffolds. Today, this burgeoning class of natural products encompasses a structurally and functionally diverse set of molecules (i.e. microcin B17, cyanobactins, and thiopeptides). TOMMs presumably provide their producers with an ecological advantage. This advantage can include chemical weapons wielded in the battle for nutrients, disease-promoting virulence factors, or compounds presumably beneficial for symbiosis. Despite this plethora of functions, many TOMMs await experimental interrogation. This review will focus on the biosynthesis and natural combinatorial diversity of the TOMM family.Graphical abstractHighlights► Thiazole/oxazole modified microcins are a widespread natural product platform. ► TOMMs display a wide range of functions, ranging from antibiotics to virulence factors. ► The inherently promiscuous biosynthetic enzymes allow for combinatorial biosynthesis. ► Bioinformatics has revealed many uncharacterized TOMMs. ► Core peptide mutations and biosynthetic expansions alter structure and function.
Co-reporter:Katie J. Molohon, Joel O. Melby, Jaeheon Lee, Bradley S. Evans, Kyle L. Dunbar, Stefanie B. Bumpus, Neil L. Kelleher, and Douglas A. Mitchell
ACS Chemical Biology 2011 Volume 6(Issue 12) pp:1307
Publication Date(Web):September 27, 2011
DOI:10.1021/cb200339d
The soil-dwelling, plant growth-promoting bacterium Bacillus amyloliquefaciens FZB42 is a prolific producer of complex natural products. Recently, a new FZB42 metabolite, plantazolicin (PZN), has been described as a member of the growing thiazole/oxazole-modified microcin (TOMM) family. TOMMs are biosynthesized from inactive, ribosomal peptides and undergo a series of cyclodehydrations, dehydrogenations, and other modifications to become bioactive natural products. Using high-resolution mass spectrometry, chemoselective modification, genetic interruptions, and other spectroscopic tools, we have determined the molecular structure of PZN. In addition to two conjugated polyazole moieties, the amino-terminus of PZN has been modified to Nα,Nα-dimethylarginine. PZN exhibited a highly selective antibiotic activity toward Bacillus anthracis, but no other tested human pathogen. By altering oxygenation levels during fermentation, PZN analogues were produced that bear variability in their heterocycle content, which yielded insight into the order of biosynthetic events. Lastly, genome-mining has revealed the existence of four additional PZN-like biosynthetic gene clusters. Given their structural uniqueness and intriguing antimicrobial specificity, the PZN class of antibiotics may hold pharmacological value.
Co-reporter:Evelyn M. Molloy, Jonathan I. Tietz, Patricia M. Blair, Douglas A. Mitchell
Bioorganic & Medicinal Chemistry (15 December 2016) Volume 24(Issue 24) pp:
Publication Date(Web):15 December 2016
DOI:10.1016/j.bmc.2016.05.021
The hygrolides, a family of 16-member-ring-containing plecomacrolides produced by Actinobacteria, exhibit numerous reported bioactivities. Using HR-MS/MS, nucleophilic 1,4-addition-based labeling, NMR, and bioinformatic analysis, we identified Streptomyces varsoviensis as a novel producer of JBIR-100, a fumarate-containing hygrolide, and elucidated the previously unknown stereochemistry of the natural product. We investigated the antimicrobial activity of JBIR-100, with preliminary insight into mode of action indicating that it perturbs the membrane of Bacillus subtilis. S. varsoviensis is known to produce compounds from multiple hygrolide sub-families, namely hygrobafilomycins (JBIR-100 and hygrobafilomycin) and bafilomycins (bafilomycin C1 and D). In light of this, we identified the biosynthetic gene cluster for JBIR-100, which, to our knowledge, represents the first reported for a hygrobafilomycin. Finally, we performed a bioinformatic analysis of the hygrolide family, describing clusters from known and predicted producers. Our results indicate that potential remains for the Actinobacteria to yield novel hygrolide congeners, perhaps with differing biological activities.
.png)
![Carbamic acid, [(1S)-3-chloro-2-oxo-1-[(phenylthio)methyl]propyl]-,phenylmethyl ester](http://img.cochemist.com/ccimg/217000/216988-02-2.png)
![Carbamic acid, [(1S)-3-chloro-2-oxo-1-[(phenylthio)methyl]propyl]-,phenylmethyl ester](http://img.cochemist.com/ccimg/217000/216988-02-2_b.png)
![(3S,4aS,8aS)-2-[(2R,3R)-3-amino-2-hydroxy-4-phenylsulfanylbutyl]-N-tert-butyl-3,4,4a,5,6,7,8,8a-octahydro-1H-isoquinoline-3-carboxamide](http://img.cochemist.com/ccimg/159900/159878-05-4.png)
![(3S,4aS,8aS)-2-[(2R,3R)-3-amino-2-hydroxy-4-phenylsulfanylbutyl]-N-tert-butyl-3,4,4a,5,6,7,8,8a-octahydro-1H-isoquinoline-3-carboxamide](http://img.cochemist.com/ccimg/159900/159878-05-4_b.png)
![Carbamic acid, [(1R)-3-diazo-2-oxo-1-[(phenylthio)methyl]propyl]-,phenylmethyl ester](http://img.cochemist.com/ccimg/159900/159878-00-9.png)
![Carbamic acid, [(1R)-3-diazo-2-oxo-1-[(phenylthio)methyl]propyl]-,phenylmethyl ester](http://img.cochemist.com/ccimg/159900/159878-00-9_b.png)

![[S-(R,S)]-Phenylmethyl [1-oxiranyl-2-(phenylthio)ethyl]carbamate](http://img.cochemist.com/ccimg/163500/163462-16-6.png)
![[S-(R,S)]-Phenylmethyl [1-oxiranyl-2-(phenylthio)ethyl]carbamate](http://img.cochemist.com/ccimg/163500/163462-16-6_b.png)





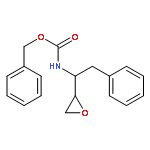
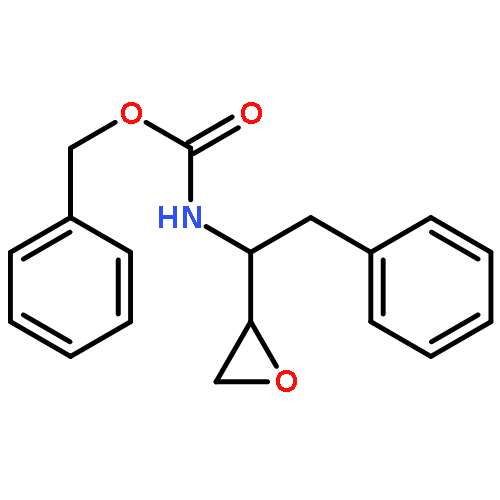



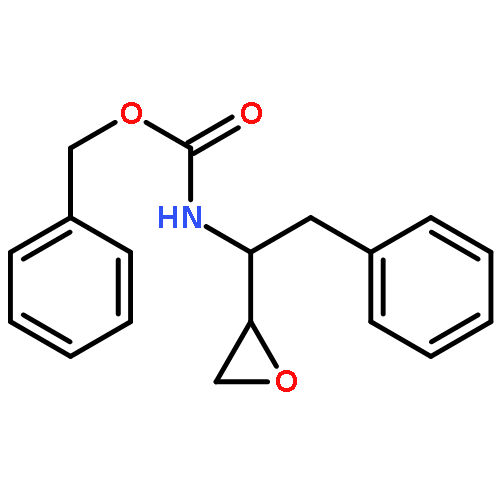

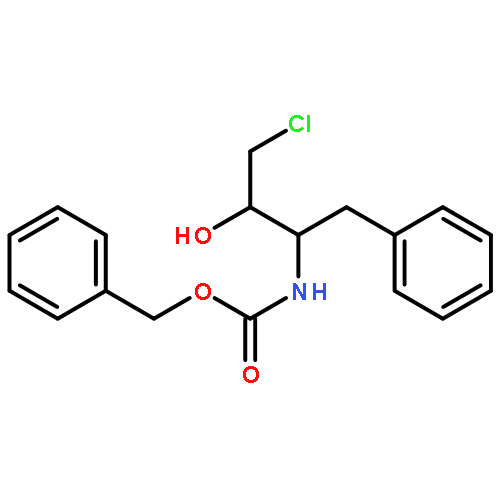


![Carbamic acid, [(1R)-3-diazo-2-oxo-1-(phenylmethyl)propyl]-, phenylmethyl ester](http://img.cochemist.com/ccimg/114700/114645-17-9.png)
![Carbamic acid, [(1R)-3-diazo-2-oxo-1-(phenylmethyl)propyl]-, phenylmethyl ester](http://img.cochemist.com/ccimg/114700/114645-17-9_b.png)








































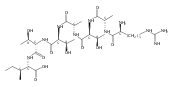


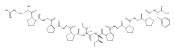

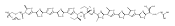
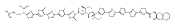





















![4-Thiazolecarboxylic acid, 2-[(2S)-2-[(4R)-2-[(4R)-4,5-dihydro-2-(2-hydroxyphenyl)-4-thiazolyl]-4-thiazolidinyl]-2-hydroxy-1,1-dimethylethyl]-4,5-dihydro-4-methyl-, (4S)-](/data/chemimg/111600/152166-40-0.png)
![4-Thiazolecarboxylic acid, 2-[(2S)-2-[(4R)-2-[(4R)-4,5-dihydro-2-(2-hydroxyphenyl)-4-thiazolyl]-4-thiazolidinyl]-2-hydroxy-1,1-dimethylethyl]-4,5-dihydro-4-methyl-, (4S)-](/data/chemimg/111600/152166-40-0_b.png)



![3H,12H-4,1:11,8:18,15-Trinitrilo-1H-pyrrolo[2,1-c][1,11,18,4,7,14,21]oxadithiatetraazacyclotetracosine-5,12,19,22(4H)-tetrone,6,7,10,11,13,14,20,21,24,25,26,26a-dodecahydro-3-methyl-7-(1-methylethyl)-14,21-bis(phenylmethyl)-,(3R,4S,7S,11R,14R,21S,26aS)- (9CI)](http://img.cochemist.com/ccimg/120900/120853-16-9.png)
![3H,12H-4,1:11,8:18,15-Trinitrilo-1H-pyrrolo[2,1-c][1,11,18,4,7,14,21]oxadithiatetraazacyclotetracosine-5,12,19,22(4H)-tetrone,6,7,10,11,13,14,20,21,24,25,26,26a-dodecahydro-3-methyl-7-(1-methylethyl)-14,21-bis(phenylmethyl)-,(3R,4S,7S,11R,14R,21S,26aS)- (9CI)](http://img.cochemist.com/ccimg/120900/120853-16-9_b.png)




![Pentanamide, 2-amino-5-[(aminoiminomethyl)amino]-, (S)-](http://img.cochemist.com/ccimg/2800/2788-83-2.png)
![Pentanamide, 2-amino-5-[(aminoiminomethyl)amino]-, (S)-](http://img.cochemist.com/ccimg/2800/2788-83-2_b.png)







