Co-reporter:Shuai Tan, Feng He, Tingting Kong, Jingde Wu, Zhaopeng Liu
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 15(Issue 15) pp:
Publication Date(Web):1 August 2017
DOI:10.1016/j.bmc.2017.05.062
As a continuous research for the discovery of coumarin-based targeted anticancer agents, we designed and synthesized a series of novel histone deacetylases (HDAC) inhibitors using the 8-ethoxy-3-nitro-2H-chromene as the surface binding or cap group, linear dicarboxylic acid or ω-amino acid moiety with different length as the linking motif, ortho-aminoanilides, amides or α-aminoamides as the zinc binding group and the internal cavity motifs. Most of these 3-nitro-2H-chromene derivatives exhibited good growth inhibitory activity against K562, A549, MCF-7, PC3 and Hela cells and were more potent than the reference drug SAHA and MS-275. At the concentration of 10 µM, the ortho-aminoanilide series and the d-Phe derived α-aminoamide derivatives 16a and 16b displayed more potent activity toward HADC1 over HADC2, and only moderate to weak activity over HADC6. In contrast, the amide ZBG analogues, 12a and 12b, 14 and 15, were only moderate HDAC6 inhibitors, but more selective over HDAC1 and HDAC2. The ortho-aminoanilides 9b, 9c, 10b, 10c, 11b, and the α-aminoamides 16a and 16b were potent HADC1 inhibitors with the IC50 values in the nanomolar ranges. The ortho-aminoanilides 10b and10c with a phenyl internal cavity motif were more potent than MS-275 as HADC1 inhibitors and more selective over HADC2.Download high-res image (83KB)Download full-size image
Co-reporter:Yan-Na Liu; Jing-Jing Wang; Ya-Ting Ji; Guo-Dong Zhao; Long-Qian Tang; Cheng-Mei Zhang; Xiu-Li Guo;Zhao-Peng Liu
Journal of Medicinal Chemistry 2016 Volume 59(Issue 11) pp:5341-5355
Publication Date(Web):May 12, 2016
DOI:10.1021/acs.jmedchem.6b00071
By targeting a new binding region at the interface between αβ-tubulin heterodimers at the colchicine binding site, we designed a series of 7-substituted 1-methyl-1,4-dihydroindeno[1,2-c]pyrazoles as potential tubulin polymerization inhibitors. Among the compounds synthesized, 2-(6-ethoxy-3-(3-ethoxyphenylamino)-1-methyl-1,4-dihydroindeno[1,2-c]pyrazol-7-yloxy)acetamide 6a and 2-(6-ethoxy-3-(3-ethoxyphenylamino)-1-methyl-1,4-dihydroindeno[1,2-c]pyrazol-7-yloxy)-N-hydroxyacetamide 6n showed noteworthy low nanomolar potency against HepG2, Hela, PC3, and MCF-7 cancer cell lines. In mechanism studies, 6a inhibited tubulin polymerization and disorganized microtubule in A549 cells by binding to tubulin colchicine binding site. 6a arrested A549 cells in G2/M phase that was related to the alterations in the expression of cyclin B1 and p-cdc2. 6a induced A549 cells apoptosis through the activation of caspase-3 and PARP. In addition, 6a inhibited capillary tube formation in a concentration-dependent manner. In nonsmall cell lung cancer xenografts mouse model, 6a suppressed tumor growth by 59.1% at a dose of 50 mg/kg (ip) without obvious toxicity, indicating its in vivo potential as anticancer agent.
Co-reporter:Yong-Li Jiang;Shui-Xian Li;Yu-Jing Liu;Lian-Ping Ge;Xiu-Zhen Han;Zhao-Peng Liu
Chemical Biology & Drug Design 2015 Volume 86( Issue 5) pp:1017-1029
Publication Date(Web):
DOI:10.1111/cbdd.12569
As a continuous research for the discovery of trehalose-based anti-invasive agents, we developed a convenient synthetic approach for the preparation of 6,6′-dideoxy-6,6′-bis(acylamino)-α,α-D-trehaloses. A series of trehalose-based amides were prepared through the trityl protection of the two primary hydroxyls of α,α-D-trehalose, benzoylation, the removal of the trityl protective group, mesylation, azidation, catalytic hydrogenation in the presence of hydrochloride, coupling reaction with a variety of acids, and subsequent debenzoylation and deacetylation in some cases. Compound 8b, 6,6′-dideoxy-6,6′-bis(2-hydroxybenzamide)-α,α-D-trehalose, was just as potent as the natural brartemicin against the invasion of murine colon 26-L5 cells. It exhibited no cytotoxicity on human breast adenocarcinoma MDA-MB-231 and murine colon 26-L5 cells. It can significantly inhibit the migration and invasion of the MDA-MB-231 cells. The anti-invasive effect of 8b was possibly related to its inhibitory activity on MMP-9, its suppression on the expression of MMP-9 and VEGF, and its deactivation of Akt.
Co-reporter:Shuai Tan ;Dr. Zhao-Peng Liu
ChemMedChem 2015 Volume 10( Issue 3) pp:441-450
Publication Date(Web):
DOI:10.1002/cmdc.201402460
Abstract
Zinc-dependent histone deacetylases (HDACs), a family of hydrolases that remove acetyl groups from lysine residues, play an important role in the regulation of multiple processes, from gene expression to protein activity. The dysregulation of HDACs is associated with many diseases including cancer, neurological disorders, cellular metabolism disorders, and inflammation. Molecules that act as HDAC inhibitors (HDACi) exhibit a variety of related bioactivities. In particular, HDACi have been applied clinically for the treatment of cancers. Inhibition through competitive binding of the catalytic domain of these enzymes has been achieved by a diverse array of small-molecule chemotypes, including a number of natural products. This review provides a systematic introduction of natural HDACi, with an emphasis on their enzyme inhibitory potency, selectivity, and biological activities, highlighting their various binding modes with HDACs.
Co-reporter:Guo-Dong Zhao, Zhao-Peng Liu
Tetrahedron 2015 Volume 71(Issue 42) pp:8033-8040
Publication Date(Web):21 October 2015
DOI:10.1016/j.tet.2015.08.055
In a reported procedure for the synthesis of the ED-71 A-ring phosphine oxide, we discovered that unusual TBS transfer occurred from the 1α-position to the sterically more constrained 2β-position, and the subsequent Michael addition of ethyl acrylate predominated at the 1α-position. The X-ray analysis of a key intermediate confirmed our observations. We made a structural revision of the reported A-ring phosphine oxide synthon and ED-71. In addition, we provided the first stereoselective synthetic approach to ED-71 A-ring phosphine oxide applying the stereochemistry of d-mannitol.The reported structures and synthesis of the A-ring phosphine oxide synthon and the drug ED-71 (eldecalcitol) in J. Am. Chem. Soc. (2001, 123, 3716–3722) are not correct. We made revisions of their structures and developed a new synthetic route to the A-ring phosphine oxide and ED-71.
Co-reporter:Chao Liu, Guo-Dong Zhao, Xinliang Mao, Tsutomu Suenaga, Toshie Fujishima, Cheng-Mei Zhang, Zhao-Peng Liu
European Journal of Medicinal Chemistry 2014 Volume 85() pp:569-575
Publication Date(Web):6 October 2014
DOI:10.1016/j.ejmech.2014.08.031
•Two new vitamin D analogues with a phenyl ring attached at C-17 to replace the C-20 and C-21 were prepared.•This modification led to almost a loss of VDR binding ability.•The two analogues maintained their antiproliferative activity.•The two analogues maintained their prodifferentiating potency.Two new analogues of the steroid hormone 1α,25-dihydroxyvitamin D3 with aromatic side chains attached at C-17 were designed to investigate their effects on VDR, HL-60 cell differentiation and tumor cell proliferation. These analogues were prepared by the classical photochemical ring opening approach. After the protection of both the 1α- and 3β-hydroxyl in 1α-hydroxydehydroepiandrosterone with TBS groups, followed by bromination with NBS and debromination in the presence of γ-collidine, the diene intermediate was obtained. Hydrazone formation followed by iodine oxidation gave a vinyl iodide. The aromatic side chain at C-17 was introduced via the Negishi coupling of the resulting intermediate with an in situ generated zinc reagent with the substituted aryl bromide (CD-side chain) in the presence of catalytic amount of Pd(PPh3)4. After the removal of the TBDMS and MOM protective groups, followed by UV irradiation and the subsequent thermal reaction, the 1α,25-(OH)2-D3 analogues with a substituted phenyl ring attached at C-17 to replace the C-20 and C-21 were prepared. In the VDR competitive binding assay, compounds 2 and 3 almost lost their binding ability, and were only 0.01% and 0.015% as potent as the 1α,25-dihydroxyvitamin D3. However, compounds 2 and 3 were as potent as 1α,25-(OH)2-D3 in inducing HL-60 cell differentiation at concentrations of 30, 100, 300, 1000 nM, respectively. Moreover, compounds 2 and 3 exhibited similar or better antiproliferative potency against MCF-7 human breast cancer cells, the IC50 values for analogues 2, 3 and the natural hormone were 7.08, 7.56, and 12.5 μM, respectively.Incorporating a phenyl ring at C-17 made the 1α,25-(OH)2-D3 analogues 2 and 3 almost lose their VDR binding ability, but maintain their antiproliferative and prodifferentiating potency.
Co-reporter:Xin Qiu, Guo-Dong Zhao, Long-Qiang Tang, Zhao-Peng Liu
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 11) pp:2465-2468
Publication Date(Web):1 June 2014
DOI:10.1016/j.bmcl.2014.04.008
The design, synthesis, and biological evaluation of a series of six HIV-1 protease inhibitors incorporating isosorbide moiety as novel P2 ligands are described. All the compounds are very potent HIV-1 protease inhibitors with IC50 values in the nanomolar or picomolar ranges (0.05–0.43 nM). Molecular docking studies revealed the formation of an extensive hydrogen-bonding network between the inhibitor and the active site. Particularly, the isosorbide-derived P2 ligand is involved in strong hydrogen bonding interactions with the backbone atoms.
Co-reporter:Shu-Qiang Yin, Min Shi, Ting-Ting Kong, Cheng-Mei Zhang, Kunkun Han, Biyin Cao, Zubin Zhang, Xiaolin Du, Long-Qian Tang, Xinliang Mao, Zhao-Peng Liu
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 11) pp:3314-3319
Publication Date(Web):1 June 2013
DOI:10.1016/j.bmcl.2013.03.097
The small chemical compound 8-ethoxy-2-(4-fluorophenyl)-3-nitro-2H-chromene (S14161) was recently identified as an inhibitor of the phosphoinositide 3-kinase (PI3K). In the present study, we designed a novel synthesis of S14161 and prepared a series of its analogues via the oxa-Michael–Henry reaction in the presence of catalytic amounts of l-proline and triethylamine. Further structural simplification led to the identification of 6-bromo-8-ethoxy-3-nitro-2H-chromene (BENC-511) that exhibited potent antiproliferative activities against a panel of 12 tumor cell lines. Compared with S14161, BENC-511 was more potent in blocking the AKT phosphorylation and inducing cancer cell apoptosis. BENC-511 also displayed more potent effects on human umbilical vein epithelial cells (HUVEC) migration, suggesting its anti-angiogenesis activity.
Co-reporter:Shu-Qiang Yin, Can-Fei Zhang, Xinliang Mao, Zhao-Peng Liu
Tetrahedron: Asymmetry 2013 Volume 24(5–6) pp:320-323
Publication Date(Web):31 March 2013
DOI:10.1016/j.tetasy.2013.02.009
Racemic 8-ethoxy-2-(4-fluorophenyl)-3-nitro-2H-chromene (S14161) was recently identified as a potent inhibitor of phosphoinositide 3-kinase (PI3K). In order to investigate the effects of its two enantiomers on tumor cell lines, we designed a novel synthesis for (R)-S14161 and (S)-S14161 using a chemical resolution and derivation strategy. The readily available 3-(tert-butyldimethylsilyloxy)-salicylaldehyde underwent a tandem oxa-Michael–Henry reaction with trans-4-fluoro-β-nitrostyrene in the presence of a catalytic amount of l-proline and triethylamine to give the 3-nitro-2H-chromene. Upon removal of the TBS protecting group, the resolution of the resulting racemic 2-(4-fluorophenyl)-8-hydroxy-3-nitro-2H-chromene was achieved via diastereomeric ester formation using (S)-(+)-α-methoxyphenylacetic acid as the derivatizing agent, followed by aminolysis. Finally, the ethyl ether formation of each enantiomer furnished (R)-S14161 and (S)-S14161 in enantiomerically pure forms. The absolute configurations of these chiral molecules were determined by a circular dichroism method. The two enantiomers showed no marked differences in inhibition of growth of human myeloma LP1 and OPM-2 cells.(2R)-2-(4-Fluorophenyl)-3-nitro-8-[(S)-α-methoxyphenylacetoxy]-2H-chromeneC24H18FNO6[α]D20=112.5 (c 0.16, CHCl3)Source of chirality: resolutionAbsolute configuration: (2R,2′S)(2S)-2-(4-Fluorophenyl)-3-nitro-8-[(S)-α-methoxyphenylacetoxy]-2H-chromeneC24H18FNO6[α]D20=+282 (c 0.11, CHCl3)Source of chirality: resolutionAbsolute configuration: (2S,2′S)(R)-2-(4-Fluorophenyl)-8-hydroxy-3-nitro-2H-chromeneC15H10FNO4[α]D20=165 (c 0.10, CHCl3)Source of chirality: resolutionAbsolute configuration: (2R)(R)-8-Ethoxy-2-(4-fluorophenyl)-3-nitro-2H-chromeneC17H14FNO4[α]D20=158 (c 0.73, CHCl3)Source of chirality: chemical derivationAbsolute configuration: (2R)
Co-reporter:Yong-Li Jiang, Satoshi Miyanaga, Xiu-Zhen Han, Long-Qiang Tang, Yasuhiro Igarashi, Ikuo Saiki and Zhao-Peng Liu
The Journal of Antibiotics 2013 66(9) pp:531-537
Publication Date(Web):May 8, 2013
DOI:10.1038/ja.2013.37
Brartemicin is a trehalose-based inhibitor of tumor cell invasion produced by the actinomycete of the genus Nonomuraea. In order to find more potent anti-invasive agents and study the structure–activity relationships, a series of 19 brartemicin analogs were prepared via two synthetic routes from α,α-D-trehalose and evaluated for their anti-invasive activities. Compound 4f, 6,6′-bis(2,3-dimethoxybenzoyl)-α,α-D-trehalose, was more potent than the natural brartemicin. It inhibited the invasion of murine colon 26-L5, colon carcinoma SW620, melanoma B16-BL6 and breast MDA-MB-231 cells with IC50 values of 0.15, 2.35, 4.12 and 2.61 μM, respectively. Analog 4p, 6,6′-bis(3,4-dimethoxycinnamoyl)-α,α-D-trehalose, was as potent as brartemicin against invasion of murine colon 26-L5 carcinoma cells in vitro. The structure–activity relationships of these novel trehalose-based compounds were summarized.
Co-reporter:Can-Fei Zhang; Ren-Zhong Wan; Zhao-Peng Liu
ChemMedChem 2013 Volume 8( Issue 8) pp:1249-1260
Publication Date(Web):
DOI:10.1002/cmdc.201300160
Abstract
The vitamin D hormone, 1α,25-dihydroxyvitamin D3 [1,25-(OH)2D3], exerts its hormonal effects predominantly on intestine, bone, and kidney, where it plays a crucial role in calcium and phosphorus homeostasis and bone mineralization. In addition to its classical actions, 1,25(OH)2D3 exerts pleiotropic effects in a wide variety of target tissues and cell types, often in an autocrine/paracrine fashion. These biological activities of 1,25(OH)2D3 have suggested a multitude of potential therapeutic applications for the vitamin D hormone in the treatment of hyperproliferative disorders (e.g. cancer and psoriasis), immune dysfunction (autoimmune diseases), and endocrine disorders (e.g. hyperparathyroidism). However, the calcemic effects induced by 1,25(OH)2D3—hypercalcemia, increased bone resorption, and soft tissue calcification—limit the use of the natural ligand in these clinical applications. Therefore, numerous 1,25(OH)2D3 analogues have been synthesized with the intent of producing therapeutic agents devoid of hypercalcemic and hyperphosphatemic side effects. To this aim, much attention has been focused on the development of 19-nor-vitamin D3 derivatives that lack the ring-A exocyclic methylene group (C19). In this review, the 19-nor-1,25(OH)2D3 analogues are classified according to modifications made at the A-ring, the side chain, or both the A-ring and side chain, as well as other positions. The biological activities of these 19-nor-1,25(OH)2D3 analogues are summarized and their structure–activity relationships and binding features with the vitamin D receptor (VDR) are discussed.
Co-reporter:Yong-Li Jiang, Long-Qian Tang, Satoshi Miyanaga, Yasuhiro Igarashi, Ikuo Saiki, Zhao-Peng Liu
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 4) pp:1089-1091
Publication Date(Web):15 February 2011
DOI:10.1016/j.bmcl.2010.12.133
Brartemicin is a trehalose-based inhibitor of tumor cell invasion produced by the actinomycete of the genus Nonomuraea. In order to explore the preliminary structure–activity relationship and obtain more potent inhibitors, a series of brartemicin analogs were synthesized through the Mitsunobu coupling of the secondary hydroxyls benzyl protected α,α-d-trehalose with benzoic acid derivatives, followed by modification of functional groups and deprotection. These compounds were evaluated for their inhibitory activity against invasion of murine colon 26-L5 carcinoma cells in vitro. Among the synthetic analogs tested, 6,6′-bis(2,3-dimethoxybenzoyl)-α,α-d-trehalose (5e) was found to be the most potent anti-invasive agent, exhibited a 2.6-fold improvement with regard to the parent natural product brartemicin, and it is considered to be a promising lead molecule for the anti-metastasis.
Co-reporter:Bing-Lei Gao, Cheng-Mei Zhang, Yi-Zhen Yin, Long-Qian Tang, Zhao-Peng Liu
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 12) pp:3730-3733
Publication Date(Web):15 June 2011
DOI:10.1016/j.bmcl.2011.04.070
A series of new HIV-1 protease inhibitors with the hydroxyethylamine core and different hydroxyprolinamide P2 ligands were designed and synthesized. Variation of substitutions at the P2 significantly affected the enzyme inhibitory potency of the inhibitors. Compounds 2a and 2d showed excellent enzyme inhibitory activity with IC50 values in the nanomolar range. An active site binding model for inhibitors 2a and 2d was suggested based upon the computational-docking results of the ligand with HIV-1 protease. This model offers molecular insights regarding ligand-binding site interactions of the hydroxyprolinamide-derived novel P2-ligand.
Co-reporter:Hui Min Sun, Zhao Peng Liu, Long Qian Tang
Chinese Chemical Letters 2008 Volume 19(Issue 8) pp:907-910
Publication Date(Web):August 2008
DOI:10.1016/j.cclet.2008.05.036
Treatment of N-t-butylbenzenesulfonamide with an excess of BuLi, followed by the reaction with methyl 2-(4-methylphenyl)propanoate, gave the corresponding 2-carboxybenzenesulfonamide, which underwent a sequence of consecutive N-deprotective cyclization process mediated by TMSCl–NaI–MeCN reagent to afford the N-sulfonylimine. Following the bromination and ring expansion, 3-methyl-3-(4-methylphenyl)-2H-benzo[e][1,2]thiazine-1,1,4-trione was obtained. Optical resolution of the racemic benzosultam using (−)-menthoxyacetyl chloride, furnished the optically pure (+)- and (−)-3-methyl-3-(4-methylphenyl)-2H-benzo[e][1,2]thiazine-1,1,4-triones, which were fluorinated with FClO3 to produce the corresponding chiral N–F agents.
Co-reporter:Zhao-Peng Liu;Zhao-Li Meng;De-Feng Wang
Chinese Journal of Chemistry 2006 Volume 24(Issue 4) pp:
Publication Date(Web):5 APR 2006
DOI:10.1002/cjoc.200690097
A novel efficient synthetic route to 1,3-dihydrobenzo[c]furan glycone was developed and the corresponding 5-fluoro, 5-iodo uracil and guanosine derivatives, the aromatic analogues of the well known antiviral 2′,3′-dideoxy-2′,3′-dihydronucleosides (d4N), were synthesized.
Co-reporter:Guo-Dong Zhao, Zhao-Peng Liu
Steroids (October 2014) Volume 88() pp:72-76
Publication Date(Web):1 October 2014
DOI:10.1016/j.steroids.2014.07.005
•We provided the (20S)- and (20R)-alcohols via the CBS stereoselective reduction.•We developed a new synthetic approach to the 22-oxa-25-hydroxy Grundmann’s ketone.•A novel synthesis of maxacalcitol is achieved by the convergent Lythgoe coupling on gram scale.Stereoselective reduction of C-20 ketone of vitamin D CD-ring precursor using Corey’s CBS reagents, (R)-(+)-2-methyl-CBS-oxazaborolidine and (S)-(−)-2-methyl-CBS-oxazaborolidine, led to the (20S)-alcohol 5 and (20R)-epimer 4 in approximately 17:1 selectivity. A new synthetic approach to the 22-oxa-25-hydroxy Grundmann’s ketone 11 was developed through the Williamson etherification of (20S)-alcohol 5 with 1-bromomethyl-2,2-dimethyloxirane, followed by regioselective reductive epoxide ring opening with LiAlH4, the removal of the silyl protecting group by TBAF, and the environmentally benign TEMPO-mediated oxidation using inexpensive Oxone as a co-oxidizing agent. The preparation of drug maxacalcitol was achieved on gram scale by the convergent Lythgoe coupling via Wittig–Horner reaction of the A-ring phosphine oxide synthon with the CD-ring fragment.Download full-size image
Co-reporter:Yi-Zhen Yin, Chao Liu, Long-Qian Tang, Zhao-Peng Liu
Steroids (November 2012) Volume 77(Issue 13) pp:1419-1422
Publication Date(Web):1 November 2012
DOI:10.1016/j.steroids.2012.08.018
A novel recyclable Pd/C catalyst mediated dehydrogenation of sterols is developed. The conversion of sterols to 1,4,6-trien-3-ones is best achieved with Pd/C as a catalyst (10%) in the presence of six equivalents of allyl diethyl phosphate (ADP) and excess amount of sodium carbonate in DMF under vigorous reflux conditions. This transformation gives 17,17-ethylenedioxyandrost-1,4,6-trien-3-one in better yield than that of DDQ oxidation and thus provides an improved synthesis of 1α-hydroxydehydroepiandrosterone from DHEA.Download full-size image
Co-reporter:Chao Liu, Fei Xie, Guo-Dong Zhao, De-Feng Wang, Hong-Xiang Lou, Zhao-Peng Liu
Steroids (December 2015) Volume 104() pp:214-219
Publication Date(Web):1 December 2015
DOI:10.1016/j.steroids.2015.10.006
•A facile synthetic route to solasodine analogues 6 and 7 from diosgenin was developed.•The 22-hydroxyfurostan analogue 4 was obtained in the opening of the spirostan F ring.•The azidation and the 22-OH dehydration were achieved in one step via Mitsunobu reaction.•An unexpected tetrahydrofuran ring opening product was identified in the Birch reduction process.The synthesis of 1α-hydroxysolasodine from diosgenin was attempted. The Pd/C catalyst mediated dehydrogenation of diosgenin generated the 1,4,6-trien-3-one (3), which was reacted with Ac2O in pyridine in the presence of a catalytic amount of POCl3 followed by hydrolysis to give the 22-hydroxyfurostan (4) in 65% yield. Conversion of the primary 26-OH group into the azide and simultaneously 22-OH dehydration were achieved in one step by Mitsunobu reaction. Treatment of the (25R)-26-azidofurosta-1,4,6,20(22)-tetraen-3-one (5) with chlorotrimethylsilane (TMSCl)/NaI/MeCN and cyclisation in situ provided the (22R,25R)-spirosola-1,4,6-trien-3-one (6) in good yield. Stereoselective and regioselective epoxidation of trienone (6) with 30% H2O2 and 5% NaOH in methanol gave the 1α,2α-epoxy-(22R,25R)-spirosola-4,6-dien-3-one (7). Birch reduction of the epoxide (7) with Li/NH3 in THF followed by the treatment with NH4Cl, however, failed to generate the expected 1α-hydroxysolasodine, but provided a tetrahydrofuran ring opening product, (22S,25R)-1α,16β-dihydroxy-22,26-epiminocholest-4-en-3-one (8). Compounds 3 and 5–8 as well as solasodine were evaluated for their cell growth inhibitory activities against human prostate cancer PC3, human cervical carcinoma Hela, and human hepatoma HepG2 cells. At the concentration of 10 μM, only epoxide 7 displayed moderate inhibitory rates towards these cells (40–54%).Download full-size image

![(2S)-2-AMINO-3-METHYL-N-[4-[3-(1,2,4-TRIAZOL-1-YL)-4-(3,4,5-TRIMETHOXYBENZOYL)PHENYL]-1,3-THIAZOL-2-YL]BUTANAMIDE;HYDROCHLORIDE](http://img.cochemist.com/data/images/1240321-53-2.png)
![(2S)-2-AMINO-3-METHYL-N-[4-[3-(1,2,4-TRIAZOL-1-YL)-4-(3,4,5-TRIMETHOXYBENZOYL)PHENYL]-1,3-THIAZOL-2-YL]BUTANAMIDE;HYDROCHLORIDE](http://img.cochemist.com/data/images/1240321-53-2_b.png)












![N-乙基-N'-[2-甲氧基-4-[5-甲基-4-[[(1S)-1-(3-吡啶基)丁基]氨基]-2-嘧啶基]苯基]脲](http://img.cochemist.com/ccimg/917200/917111-44-5.png)
![N-乙基-N'-[2-甲氧基-4-[5-甲基-4-[[(1S)-1-(3-吡啶基)丁基]氨基]-2-嘧啶基]苯基]脲](http://img.cochemist.com/ccimg/917200/917111-44-5_b.png)
![{(1R,2R)-2-[(2Z,4S)-4-HYDROXY-2-PENTEN-1-YL]-3-OXOCYCLOPENTYL}ACE<WBR />TIC ACID](http://img.cochemist.com/ccimg/714300/714272-27-2.png)
![{(1R,2R)-2-[(2Z,4S)-4-HYDROXY-2-PENTEN-1-YL]-3-OXOCYCLOPENTYL}ACE<WBR />TIC ACID](http://img.cochemist.com/ccimg/714300/714272-27-2_b.png)
![2-AMINO-2-(HYDROXYMETHYL)PROPANE-1,3-DIOL;[2-METHOXY-5-[(Z)-2-(3,4,5-TRIMETHOXYPHENYL)ETHENYL]PHENYL] DIHYDROGEN PHOSPHATE](/data/chemimg/59100/404886-32-4.png)
![2-AMINO-2-(HYDROXYMETHYL)PROPANE-1,3-DIOL;[2-METHOXY-5-[(Z)-2-(3,4,5-TRIMETHOXYPHENYL)ETHENYL]PHENYL] DIHYDROGEN PHOSPHATE](/data/chemimg/59100/404886-32-4_b.png)
![[3R-(1Z,3β,4α,5α)]-2-[3,5-bis[(1,1-dimethylethyl)dimethylsilyloxy]-4-hydroxy-2-methylenecyclohexylidene]ethyl trimethylacetate](http://img.cochemist.com/ccimg/342700/342644-90-0.png)
![[3R-(1Z,3β,4α,5α)]-2-[3,5-bis[(1,1-dimethylethyl)dimethylsilyloxy]-4-hydroxy-2-methylenecyclohexylidene]ethyl trimethylacetate](http://img.cochemist.com/ccimg/342700/342644-90-0_b.png)


![2-AMINO-3-HYDROXY-N-[2-METHOXY-5-[2-(3,4,5-TRIMETHOXYPHENYL)ETHENYL]PHENYL]PROPANAMIDE;HYDROCHLORIDE](http://img.cochemist.com/ccimg/253500/253426-24-3.png)
![2-AMINO-3-HYDROXY-N-[2-METHOXY-5-[2-(3,4,5-TRIMETHOXYPHENYL)ETHENYL]PHENYL]PROPANAMIDE;HYDROCHLORIDE](http://img.cochemist.com/ccimg/253500/253426-24-3_b.png)
![Benzenesulfonamide,N-[(2R,3S)-3-amino-2-hydroxy-4-phenylbutyl]-N-(2-methylpropyl)-4-nitro-](http://img.cochemist.com/ccimg/251200/251105-80-3.png)
![Benzenesulfonamide,N-[(2R,3S)-3-amino-2-hydroxy-4-phenylbutyl]-N-(2-methylpropyl)-4-nitro-](http://img.cochemist.com/ccimg/251200/251105-80-3_b.png)


![(5s)-5-acetamido-9,10,11-trimethoxy-6,7-dihydro-5h-dibenzo[a,c][7 ]annulen-3-yl Dihydrogen Phosphate](http://img.cochemist.com/ccimg/220000/219923-05-4.png)
![(5s)-5-acetamido-9,10,11-trimethoxy-6,7-dihydro-5h-dibenzo[a,c][7 ]annulen-3-yl Dihydrogen Phosphate](http://img.cochemist.com/ccimg/220000/219923-05-4_b.png)
![(3r,3ar,6s,6as)-6-[tert-butyl(dimethyl)silyl]oxy-2,3,3a,5,6,6a-he Xahydrofuro[3,2-b]furan-3-ol](http://img.cochemist.com/ccimg/205000/204909-70-6.png)
![(3r,3ar,6s,6as)-6-[tert-butyl(dimethyl)silyl]oxy-2,3,3a,5,6,6a-he Xahydrofuro[3,2-b]furan-3-ol](http://img.cochemist.com/ccimg/205000/204909-70-6_b.png)


![disodium,[2-methoxy-5-[(Z)-2-(3,4,5-trimethoxyphenyl)ethenyl]phenyl] phosphate](http://img.cochemist.com/ccimg/168600/168555-66-6.png)
![disodium,[2-methoxy-5-[(Z)-2-(3,4,5-trimethoxyphenyl)ethenyl]phenyl] phosphate](http://img.cochemist.com/ccimg/168600/168555-66-6_b.png)


![Benzenesulfonamide,N-[2-[(4-hydroxyphenyl)amino]-3-pyridinyl]-4-methoxy-](http://img.cochemist.com/ccimg/141500/141430-65-1.png)
![Benzenesulfonamide,N-[2-[(4-hydroxyphenyl)amino]-3-pyridinyl]-4-methoxy-](http://img.cochemist.com/ccimg/141500/141430-65-1_b.png)
![(1s,4s,7z,10s,16e,21r)-7-ethylidene-4,21-di(propan-2-yl)-2-oxa-12,13-dithia-5,8,20,23-tetrazabicyclo[8.7.6]tricos-16-ene-3,6,9,19,22-pentone](http://img.cochemist.com/ccimg/128600/128517-07-7.png)
![(1s,4s,7z,10s,16e,21r)-7-ethylidene-4,21-di(propan-2-yl)-2-oxa-12,13-dithia-5,8,20,23-tetrazabicyclo[8.7.6]tricos-16-ene-3,6,9,19,22-pentone](http://img.cochemist.com/ccimg/128600/128517-07-7_b.png)
![1,3-Cyclohexanediol,2-(3-hydroxypropoxy)-4-methylene-5-[(2E)-2-[(1R,3aS,7aR)-octahydro-1-[(1R)-5-hydroxy-1,5-dimethylhexyl]-7a-methyl-4H-inden-4-ylidene]ethylidene]-,(1R,2R,3R,5Z)-](http://img.cochemist.com/ccimg/104200/104121-92-8.png)
![1,3-Cyclohexanediol,2-(3-hydroxypropoxy)-4-methylene-5-[(2E)-2-[(1R,3aS,7aR)-octahydro-1-[(1R)-5-hydroxy-1,5-dimethylhexyl]-7a-methyl-4H-inden-4-ylidene]ethylidene]-,(1R,2R,3R,5Z)-](http://img.cochemist.com/ccimg/104200/104121-92-8_b.png)
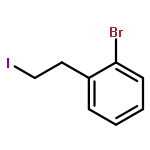
![Propanedioic acid, [2-(2-bromophenyl)ethyl]-](http://img.cochemist.com/ccimg/92100/92013-29-1.png)
![Propanedioic acid, [2-(2-bromophenyl)ethyl]-](http://img.cochemist.com/ccimg/92100/92013-29-1_b.png)
![[3S-(1Z,3a,5b)]-[2-[3,5-Bis[[(tert-butyl)dimethylsilyl]oxy]-2-methylenecyclohexylidene]ethyl]diphenylphosphineoxide](http://img.cochemist.com/ccimg/81600/81522-68-1.png)
![[3S-(1Z,3a,5b)]-[2-[3,5-Bis[[(tert-butyl)dimethylsilyl]oxy]-2-methylenecyclohexylidene]ethyl]diphenylphosphineoxide](http://img.cochemist.com/ccimg/81600/81522-68-1_b.png)
![[(2S)-2-[(1R,3aR)-4-hydroxy-7a-methyl-1,2,3,3a,4,5,6,7-octahydroinden-1-yl]propyl] 4-methylbenzenesulfonate](http://img.cochemist.com/ccimg/66800/66774-80-9.png)
![[(2S)-2-[(1R,3aR)-4-hydroxy-7a-methyl-1,2,3,3a,4,5,6,7-octahydroinden-1-yl]propyl] 4-methylbenzenesulfonate](http://img.cochemist.com/ccimg/66800/66774-80-9_b.png)
![(1r,3s,5z)-5-[(2e)-2-[(1r,3as,7ar)-1-[(2r)-6-hydroxy-6-methylheptan-2-yl]-7a-methyl-2,3,3a,5,6,7-hexahydro-1h-inden-4-ylidene]ethylidene]-4-methylidenecyclohexane-1,3-diol](http://img.cochemist.com/ccimg/32300/32222-06-3.png)
![(1r,3s,5z)-5-[(2e)-2-[(1r,3as,7ar)-1-[(2r)-6-hydroxy-6-methylheptan-2-yl]-7a-methyl-2,3,3a,5,6,7-hexahydro-1h-inden-4-ylidene]ethylidene]-4-methylidenecyclohexane-1,3-diol](http://img.cochemist.com/ccimg/32300/32222-06-3_b.png)
![Benzene, 1-methoxy-4-[(2-propynyloxy)methyl]-](http://img.cochemist.com/ccimg/4100/4039-83-2.png)
![Benzene, 1-methoxy-4-[(2-propynyloxy)methyl]-](http://img.cochemist.com/ccimg/4100/4039-83-2_b.png)





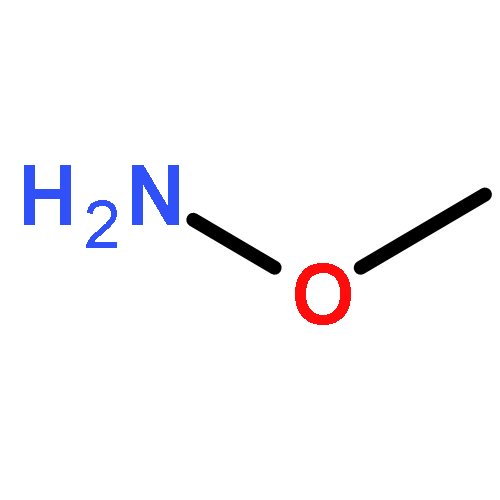
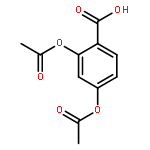
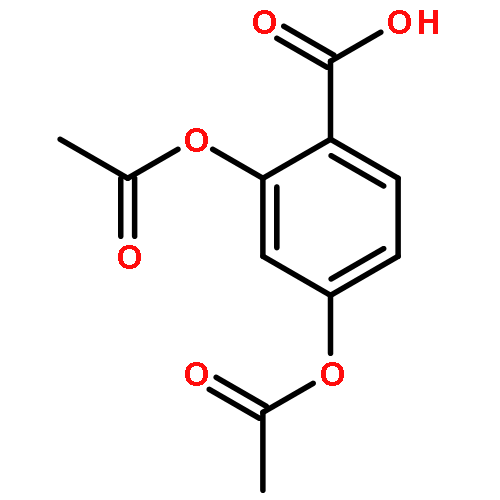
![(1R,3S,5Z)-5-[(2E)-2-[(1S,3AS,7AS)-1-[(1S)-1-(3-HYDROXY-3-METHYLBUTOXY)ETHYL]-7A-METHYL-2,3,3A,5,6,7-HEXAHYDRO-1H-INDEN-4-YLIDENE]ETHYLIDENE]-4-METHYLIDENECYCLOHEXANE-1,3-DIOL](http://img.cochemist.com/ccimg/104000/103909-75-7.png)
![(1R,3S,5Z)-5-[(2E)-2-[(1S,3AS,7AS)-1-[(1S)-1-(3-HYDROXY-3-METHYLBUTOXY)ETHYL]-7A-METHYL-2,3,3A,5,6,7-HEXAHYDRO-1H-INDEN-4-YLIDENE]ETHYLIDENE]-4-METHYLIDENECYCLOHEXANE-1,3-DIOL](http://img.cochemist.com/ccimg/104000/103909-75-7_b.png)