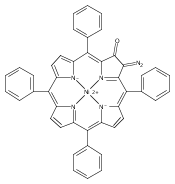
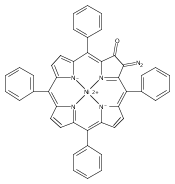
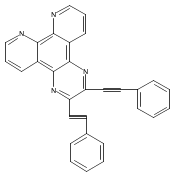
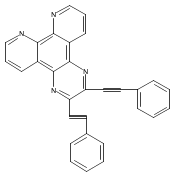
Co-reporter:Changsheng Shan, Erin T. Martin, Dennis G. Peters, and Jeffrey M. Zaleski
Chemistry of Materials July 25, 2017 Volume 29(Issue 14) pp:6030-6030
Publication Date(Web):June 26, 2017
DOI:10.1021/acs.chemmater.7b01813
Environmental impacts of continued CO2 production have led to an increased need for new methods of CO2 removal and energy development. Nanomaterials are of special interest for these applications, because of their unique chemical and physical properties that allow for highly active surfaces. Here, we successfully synthesize AgPd nanodendrite-modified Au nanoprisms in various shapes (nanoprisms, hexagonal nanoplates, and octahedral nanoparticles) by selective metal deposition. This strategy involves coupling galvanic replacement between Ag layers in Au@Ag core–shell nanoprisms and H2PdCl4 with a coreduction process of silver and palladium ions. Synthesis of AgPd nanodendrite-tipped (4.14–11.47 wt % Pd) and -edged (25.25–31.01 wt % Pd) Au nanoparticles can be controlled simply by tuning the concentration of H2PdCl4. More importantly, these multicomponent AgPd nanodendrite-modified Au nanoparticles show exceptional electrocatalytic performance for CO2 reduction. AgPd nanodendrite-edged Au nanoprisms show more favorable potentials (−0.18 V vs RHE) than previously reported nanocatalysts for the reduction of CO2 to formate, and exhibit higher faradaic efficiencies (49%) than Au, Au@Ag, and AgPd nanodendrite-tipped Au nanoprisms in aqueous electrolytes. Moreover, AgPd nanodendrite-modified Au nanoprisms show much higher selectivity and faradaic efficiency for CO2 reduction to CO (85–87%) than Au and Au@Ag nanoprisms (43–64%) in organic electrolytes. The high performance of these particles for CO2 reduction is attributed to the unique structure of AgPd nanodendrite-modified Au nanoprisms and the synergistic effect of Ag having an affinity for CO2, efficient binding of hydrogen at Pd, and Au as a stable, conductive support. In addition, AgPd nanodendrite-edged Au nanoprisms show highly stable catalytic activity during long-term electrolyses (up to 12 h) and repetitive use. These exciting results indicate that AgPd nanodendrite-modified Au nanoparticles are promising for application in CO2 conversion into useful fuels.
Co-reporter:Joan M. Walker and Jeffrey M. Zaleski
Nanoscale 2016 vol. 8(Issue 3) pp:1535-1544
Publication Date(Web):15 Dec 2015
DOI:10.1039/C5NR06700F
Developing facile synthetic routes to multifunctional nanoparticles combining the magnetic properties of iron oxides with the optical and catalytic utility of noble metal particles remains an important goal in realizing the potential of hybrid nanomaterials. To this end, we have developed a single route to noble metal-decorated magnetic nanoparticles (Fe3O4@SiO2–M; M = Au, Pd, Ag, and PtAg) and characterized them by HRTEM and STEM/EDX imaging to reveal their nanometer size (16 nm Fe3O4 and 1–5 nm M seeds) and uniformity. This represents one of the few examples of genuine multifunctional particles on the nanoscale. We show that these hybrid structures have excellent catalytic activity for the reduction of 4-nitrophenol (knorm = 2 × 107 s−1 mol(Pd)−1; 5 × 106 s−1 mol(Au)−1; 5 × 105 s−1 mol(PtAg)−1; 7 × 105 s−1 mol(Ag)−1). These rates are the highest reported for nano-sized comparables, and are competitive with mesoparticles of similar composition. Due to their magnetic response, the particles are also suitable for magnetic recovery and maintain >99% conversion for at least four cycles. Using this synthetic route, Fe3O4@SiO2–M particles show great promise for further development as a precursor to complicated anisotropic materials or for applications ranging from nanocatalysis to biomedical sensing.
Co-reporter:Jordan A. Harrington, Zachary D. Harms and Jeffrey M. Zaleski
RSC Advances 2016 vol. 6(Issue 64) pp:59113-59123
Publication Date(Web):13 Jun 2016
DOI:10.1039/C6RA09613A
Herein we adhere gold nanorods (Au NRs) to glass coverslips through electrostatic interactions by treating the glass coverslips with (3-aminopropyl)trimethoxysilane (APTMS) and polystyrene sulfonate (PSS). These Au NR-functionalized coverslips were used to demonstrate the sustainable, catalytic reduction of 4-nitroaniline by sodium borohydride. Au NRs in solution were compared to the Au NR coverslips and were found to have pseudo-first-order observed rate constants of 1.35 × 10−2 min−1 (2.79 × 106 min−1 per Au) and 4.12 × 10−2 min−1 (4.26 × 105 min−1 per Au), respectively. The same catalytic reaction was executed under photolysis (1000 W Xe lamp); free NRs and Au NR-functionalized coverslips demonstrate pseudo-first-order observed rate constants of 2.15 × 10−2 min−1 (4.44 × 106 min−1 per Au) and 6.28 × 10−2 min−1 (6.48 × 105 min−1 per Au), respectively, which are two orders of magnitude faster than the only other affixed catalyst, Au NP in polymer. In addition to being scalable in Au-NR layers, a representative Au NR-functionalized coverslip was re-used for three consecutive catalysis reactions before decrease in rate, and had a rate ∼65% of the original reaction after the fifth cycle. Thus, we have demonstrated an efficient and reusable, thermal and photochemical gold nanorod catalyst assembly that does not require a complex synthetic protocol or the use of specialized equipment.
Co-reporter:Meghan R. Porter, Jeffrey M. Zaleski
Polyhedron 2016 Volume 103(Part A) pp:187-195
Publication Date(Web):8 January 2016
DOI:10.1016/j.poly.2015.10.041
One of the key concerns with the development of radical-generating reactive therapeutics is the ability to control the activation event within a biological environment. To that end, a series of quinoline–metalloenediynes of the form M(QuiED)·2Cl (M = Cu(II), Fe(II), Mg(II), or Zn(II)) and their independently synthesized cyclized analogs have been prepared in an effort to elucidate Bergman cyclization (BC) reactivity differences in solution. HRMS(ESI) establishes a solution stoichiometry of 1:1 metal to ligand with coordination of one chloride counter ion to the metal center. EPR spectroscopy of Cu(QuiED)·2Cl and Cu(QuiBD)·2Cl denotes an axially-elongated tetragonal octahedron (g|| > g⊥ > 2.0023) with a dx2-y2dx2-y2 ground state, while the electronic absorption spectrum reveals a pπ Cl→Cu(II) LMCT feature at 19,000 cm−1, indicating a solution structure with three nitrogens and a chloride in the equatorial plane with the remaining quinoline nitrogen and solvent in the axial positions. Investigations into the BC activity reveal formation of the cyclized product from the Cu(II) and Fe(II) complexes after 12 h at 45 °C in solution, while no product is observed for the Mg(II) or Zn(II) complexes under identical conditions. The basis of this reactivity difference has been found to be a steric effect leading to metal–ligand bond elongation and thus, a retardation of solution reactivity. These results demonstrate how careful consideration of ligand and complex structure may allow for a degree of control and selective activation of these reactive agents.A series of metalloenediynes containing Fe(II), Cu(II), Zn(II), or Mg(II) have been prepared which demonstrate differing solution Bergman cyclization relativities. EPR and UV–Vis spectroscopic investigations reveal an interesting binding mode of the tetradentate ligand. Our results demonstrate how complex structure may allow for selective activation of these reactive agents.
Co-reporter:Joan M. Walker and Jeffrey M. Zaleski
Chemistry of Materials 2015 Volume 27(Issue 24) pp:8448
Publication Date(Web):November 24, 2015
DOI:10.1021/acs.chemmater.5b04134
Halting cancer progression by altering the tumor microenvironment is an exciting new frontier in oncology. One target for new therapies is the structural support provided by the extracellular matrix, which for cancer cells is often abnormal and undergoes drastic modification during angiogenesis, tumor proliferation, and metastasis. We have developed a new magnetic nanoparticle-based agent, Fe3O4–PEG–EDDA (EDDA: (Z)-octa-4-en-2,6-diyne-1,8-diamine), that is capable of generating radicals via Bergman cyclization of the pendant enediyne during mild hyperthermia treatment using an alternating magnetic field. We observe formation of a cyclized aromatic product in the Raman spectra of the thermally excited material and can detect polymeric product after periods of induction. When mixed with the basement membrane extract Matrigel, the nanoparticles do not interfere with normal biopolymer network formation. Applying the same hyperthermia conditions as in solution samples, Fe3O4–PEG–EDDA causes structural collapse of the matrix, visible by electron microscopy, in contrast to the nonradical-forming thermal control Fe3O4–PEG–BD (BD: o-xylylenediamine). Localized damage to the extracellular matrix by nanoparticle-induced molecular transformations represent a conceptual new tool in tumor microenvironment modification.
Co-reporter:Meghan R. Porter, Akiko Kochi, Jonathan A. Karty, Mi Hee Lim and Jeffrey M. Zaleski
Chemical Science 2015 vol. 6(Issue 2) pp:1018-1026
Publication Date(Web):30 Oct 2014
DOI:10.1039/C4SC01979B
Current approaches toward modulation of metal-induced Aβ aggregation pathways involve the development of small molecules that bind metal ions, such as Cu(II) and Zn(II), and interact with Aβ. For this effort, we present the enediyne-containing ligand (Z)-N,N′-bis[1-pyridin-2-yl-meth(E)-ylidene]oct-4-ene-2,6-diyne-1,8-diamine (PyED), which upon chelation of Cu(II) and Zn(II) undergoes Bergman-cyclization to yield diradical formation. The ability of this chelation-triggered diradical to modulate Aβ aggregation is evaluated relative to the non-radical generating control pyridine-2-ylmethyl-(2-{[(pyridine-2-ylmethylene)-amino]-methyl}-benzyl)-amine (PyBD). Variable-pH, ligand UV-vis titrations reveal pKa = 3.81(2) for PyBD, indicating it exists mainly in the neutral form at experimental pH. Lipinski's rule parameters and evaluation of blood–brain barrier (BBB) penetration potential by the PAMPA–BBB assay suggest that PyED may be CNS+ and penetrate the BBB. Both PyED and PyBD bind Zn(II) and Cu(II) as illustrated by bathochromic shifts of their UV-vis features. Speciation diagrams indicate that Cu(II)–PyBD is the major species at pH 6.6 with a nanomolar Kd, suggesting the ligand may be capable of interacting with Cu(II)–Aβ species. In the presence of Aβ40/42 under hyperthermic conditions (43 °C), the radical-generating PyED demonstrates markedly enhanced activity (2–24 h) toward the modulation of Aβ species as determined by gel electrophoresis. Correspondingly, transmission electron microscopy images of these samples show distinct morphological changes to the fibril structure that are most prominent for Cu(II)–Aβ cases. The loss of CO2 from the metal binding region of Aβ in MALDI-TOF mass spectra further suggests that metal–ligand–Aβ interaction with subsequent radical formation may play a role in the aggregation pathway modulation.
Co-reporter:Joan M. Walker and Jeffrey M. Zaleski
Chemistry of Materials 2014 Volume 26(Issue 17) pp:5120
Publication Date(Web):August 21, 2014
DOI:10.1021/cm5024713
Thrombosis is a hallmark of several chronic diseases leading to potentially fatal heart attacks and strokes. Frontline interventions include intravenous delivery of potent, enzymatic fibrinolytics that possess a high risk for inducing systemic bleeding. As a conceptual countermeasure, we have developed a water-soluble PEGylated gold nanoparticle appended with the enediyne diamine (Z)-octa-4-en-2,6-diyne-1,8-diamine that is capable of photothermally generating 1,4-diradical species under visible excitation (λ = 514 nm, 100 mW, 2–6 h). In the absence of biopolymer substrate, photothermal excitation of these particles leads to self-quenching polymer coating formation in water. When these radical-generating nanoparticles are intrinsically applied toward the blood clot structural protein assembly fibrin, as well as its nonpolymerized precursor protein fibrinogen, scanning electron microscopy images reveal significantly modified fibrin clot morphology, as evidenced by larger void spaces and collapsed fiber regions. Quantitatively, laser confocal microscopy images of Alexa Fluor 488-labeled fibrin clots extrinsically treated with nanoparticles at the clot/solution interface show that photothermal radical formation by these particles leads to marked increase in the number of larger pore sizes (>2.0 μm) within the fibrin matrix, which derive from a corresponding decrease in the histogram of smaller pore sizes (1.5–2.0 μm). These larger pore sizes ultimately result in total perfusion of solution through the entire clot volume. The chemical manifestation of this is that radical-induced modifications occur mainly at the protein level but lead to morphological changes at the micron scale. Overall, this technology could have significant impact for disease states such as deep vein thrombosis via a localized, catheter-delivered approach.
Co-reporter:Leigh J.K. Boerner, David. F. Dye, Tillmann Köpke, Jeffrey M. Zaleski
Coordination Chemistry Reviews 2013 Volume 257(Issue 2) pp:599-620
Publication Date(Web):15 January 2013
DOI:10.1016/j.ccr.2012.07.009
Porphyrins have remarkable utility in optical and materials applications due to their extensive π-network and the corresponding electronic properties associated with this delocalization. Recent strategies have focused on methods to modulate these properties by chemically extending the conjugation. Our approach to periphery modification has centered on the application of diradicals generated through unique diyne-ene and diazo-keto functionalization at the β,β′-position of the macrocycle. In the former case, Bergman cyclization of the enediyne motif generates a new aromatic ring via a 1,4-phenyl diradical intermediate. In the absence of bimolecular quenchers in very high concentrations, the diradical adds across the adjacent phenyl rings of the meso-positions to extend the aromaticity across three rings. In the second case, dione or diazo-keto functionalization and subsequent reaction with nucleophiles in the presence of Ag+ generates acetals that lead to modified chemical properties and potentially, additional reactivity at the periphery. Diazoketochlorin photolysis on the other hand, leads to rapid N2 extrusion and initial carbene formation. The carbene is short lived and cannot be trapped even at extremely low temperatures (2 K). The subsequent Wolff ring-contracted ketene, however, is detectable, demonstrating that the out-of-plane electronic configuration is initially generated and reacts to give the nucleophile-quenched species, as well as relaxing to form exocyclic ring addition and other radical-based products. In addition to generating unique infrared and electronic spectral properties, together these strategies offer unusual synthetic opportunities, to markedly modulate macrocycle electronic structure in a highly asymmetric manner to form truly distinct porphyrinoid constructs.Graphical abstractHighlights► Approach to periphery modification centered on the application of diradicals generated through unique diyne-ene and diazo-keto functionalization at the β,β′-position of the macrocycle. ► Diradical adds across the adjacent phenyl rings of the meso-positions to extend the aromaticity across three rings. ► Dione or diazo-keto functionalization and subsequent reaction with nucleophiles in the presence of Ag+, generates acetals that lead to modified chemical properties additional reactivity potential at the periphery – diazoketochlorin photolysis leads to rapid N2 extrusion and initial carbene formation. ► Wolff ring-contracted ketene, is detectable, demonstrating that the out-of-plane electronic configuration is initially generated and reacts to give the nucleophile-quenched species, as well as relaxing to form exocyclic ring addition and other radical-based products. ► Synthetic opportunities to markedly modulate macrocycle electronic structure in a highly asymmetric manner to form truly distinct porphyrinoid constructs.
Co-reporter:Sarah E. Lindahl ; Hyunsoo Park ; Maren Pink
Journal of the American Chemical Society 2013 Volume 135(Issue 10) pp:3826-3833
Publication Date(Web):February 22, 2013
DOI:10.1021/ja308190q
Reaction of 2 equiv of 1,2-bis((diphenylphosphino)ethynyl)benzene (dppeb, 1) with Pt(cod)Cl2 followed by treatment with N2H4 yields the reduced Pt(0) metalloenediyne, Pt(dppeb)2, 2. This complex is stable to both air oxidation and metal-mediated Bergman cyclization under ambient conditions due to the nearly idealized tetrahedral geometry. Reaction of 2 with 1 equiv of I2 in the presence of excess 1,4-cyclohexadiene (1,4-CHD) radical trap rapidly and near-quantitatively generates the cis-Bergman-cyclized, diiodo product 3 (31P: δ = 41 ppm, JPt–P = 3346 Hz) with concomitant loss of 1 equiv of uncyclized phosphine chelate (31P: δ = −33 ppm). In contrast, addition of 2 equiv of I2 in the absence of additional radical trap instantaneously forms a metastable Pt(dppeb)22+ intermediate species, 4, that is characterized by δ = 51 ppm in the 31P NMR (JPt–P = 3171 Hz) and νC≡C = 2169 cm–1 in the Raman profile, indicating that it is an uncyclized, bis-ligated complex. Over 24 h, 4 undergoes ligand exchange to form a neutral, square planar complex that spontaneously Bergman cyclizes at ambient temperature to give the crystalline product Pt(dppnap-I2)I2 (dppnap-I2 = (1,4-diiodonaphthalene-2,3-diyl)bis(diphenylphosphine)), 5, in 52% isolated yield. Computational analysis of the oxidation reaction proposes two plausible flattened tetrahedral structures for intermediate 4: one where the phosphine core has migrated to a trans-spanning chelate geometry, and a second, higher energy structure (3.3 kcal/mol) with two cis-chelating phosphine ligands (41° dihedral angle) via a restricted alkyne-terminal starting point. While the energies are disparate, the common theme in both structures is the elongated Pt–P bond lengths (>2.4 Å), indicating that nucleophilic ligand substitution by I– is on the reaction trajectory to the cyclized product 5. The efficiency of the redox-mediated Bergman cyclization reaction of this stable Pt(0) metalloenediyne prodrug and resulting cisplatin-like byproduct represents an intriguing new strategy for potential dual-threat metalloenediyne therapeutics.
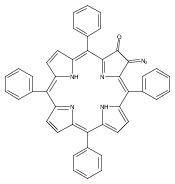
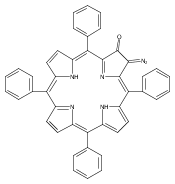
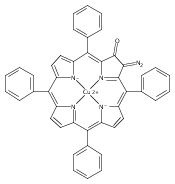
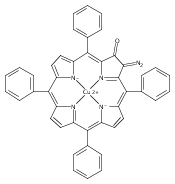
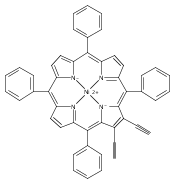
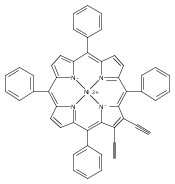
Co-reporter:Leigh J. K. Boerner, Maren Pink, Hyunsoo Park, Amanda LeSueur and Jeffrey M. Zaleski
Chemical Communications 2013 vol. 49(Issue 21) pp:2145-2147
Publication Date(Web):29 Jan 2013
DOI:10.1039/C3CC34471A
Thermal Bergman cyclization of Pt(II) dialkynylporphyrins reveals a marked reduction in the cyclization temperature relative to the free base and Zn(II) derivatives. In contrast, photogenerated 3ππ* population produces no detectable Bergman photocyclization, suggesting that the photoreactivities of the related free base and Zn(II) derivatives occurs via the 1ππ* state.
Co-reporter:David F. Dye, Tillmann Köpke, Raghunath O. Ramabhadran, Krishnan Raghavachari, and Jeffrey M. Zaleski
Journal of the American Chemical Society 2011 Volume 133(Issue 33) pp:13110-13120
Publication Date(Web):June 30, 2011
DOI:10.1021/ja203451k
Photolysis of metalated (Cu and Ni) and free base 2-diazo-3-oxochlorins within a frozen matrix (λ = 457.9 nm, toluene, 80 K) generates a single photointermediate with a hypsochromically shifted electronic absorption spectrum relative to the starting diazochlorins. The appearance of ketene (∼2131 cm–1) and azete (∼1670 cm–1) vibrations in infrared absorption and Raman spectra, respectively, identifies this intermediate as resulting from the Wolff rearrangement of the diazochlorins upon N2 loss. Computational modeling of the vibrational spectra and TDDFT simulation of the electronic transitions of potential photointermediates corroborate this assignment. Isolation and analysis of photoproducts of these diazochlorins formed within n-butanol-doped frozen toluene matrices indicate near exclusive formation of azeteoporphyrins. In sharp contrast, room temperature laser photolysis of these materials yields a mixture of photoproducts deriving from the presence of both carbene and ketene intermediates. Computational modeling of the intramolecular reactivity of the proposed sp2 carbene intermediate shows exclusive bond insertion to the adjacent phenyl group, and no evidence of Wolff rearrangement. Computational reaction profile analyses reveal that the barrierless Wolff rearrangement proceeds via an out-of-plane carbene electronic configuration that is generated directly during the loss of N2. The formation of out-of-plane carbene, resulting in the exclusive formation of the observed ketene photointermediate at low temperatures, is consistent with orbital symmetry considerations and by the geometric constraints imposed by the frozen matrix. Combined, this leads to a model showing that azeteoporphyrin formation via the Wolff rearrangement is dependent upon the structural disposition of the adjacent framework, and the specific reaction intermediate formed is very sensitive to this feature.
Co-reporter:Joan M. Walker, Linfeng Gou, Sibaprasad Bhattacharyya, Sarah E. Lindahl, and Jeffrey M. Zaleski
Chemistry of Materials 2011 Volume 23(Issue 23) pp:5275
Publication Date(Web):November 10, 2011
DOI:10.1021/cm202741p
Gold nanoparticle theranostic agents have dramatic potential in the fight against disease, particularly cancer, as multifunctional platforms combining biocompatibility, unique optical properties for detection/activation, and heat generation. In this vein, a new thiol-functionalized enediyne surfactant ligand was synthesized and coordinated to gold nanoparticles, as confirmed by the red-shift in the optical spectrum from λ = 520 to 529 nm upon ligand exchange. Raman spectra of the nanoparticle conjugate material show characteristic vibrations at 2192 (alkyne), 1582 (alkene), and 670 cm–1 (C–S). The photoreactivity of the material is explored under two sets of photolysis conditions: solution, λexc = 514 nm, RT, t = 8 h; solid aggregate, λexc = 785 nm, T = −190 °C, t = 4 h. Under these conditions, exciting into the surface plasmon of the Au nanoparticle substrate transfers heat to the organic ligand layer, initiating enediyne cyclization and generating surface radicals that lead to subsequent polymerization. New vibrational signatures arise in the alkyne (2170–1900 cm–1) and aromatic (1520–1200 cm–1) spectral regions, indicating the formation of highly conjugated species in the initial stages of the photoreaction. Prolonged irradiation results in the observation of a dense polymer coating in the TEM images, complete loss of observable molecular vibrations in the Raman spectra as a result of strong fluorescence, and a red-shift and broadening of the surface plasmon band in the electronic spectrum. Translation of this approach to nanorods and other architectures is also possible with carbon coatings clearly visible by TEM. The reported nanomaterial design represents a new approach to developing reactive biomedical agents for phototherapy applications, as well as a novel method toward carbonaceous coatings of nanoarchitectures.Keywords: (Au nanoparticles; photothermal activation; radical polymerization); surface functionalization;
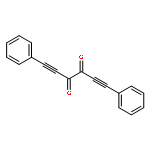
Co-reporter:Mahendra Nath, Maren Pink, Jeffrey M. Zaleski
Journal of Organometallic Chemistry 2011 696(25) pp: 4152-4157
Publication Date(Web):
DOI:10.1016/j.jorganchem.2011.07.008
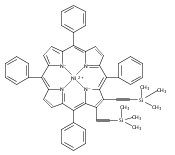
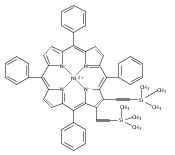
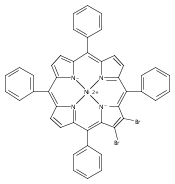
Co-reporter:Leigh J. K. Boerner;Dr. Mahendra Nath;Dr. Maren Pink; Jeffrey M. Zaleski
Chemistry - A European Journal 2011 Volume 17( Issue 34) pp:9311-9315
Publication Date(Web):
DOI:10.1002/chem.201101741
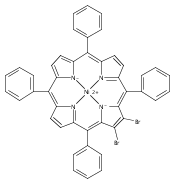
Co-reporter:Dr. Leigh J. K. Boerner;Shivnath Mazumder;Dr. Maren Pink; Mu-Hyun Baik; Jeffrey M. Zaleski
Chemistry - A European Journal 2011 Volume 17( Issue 51) pp:14539-14551
Publication Date(Web):
DOI:10.1002/chem.201102488
Abstract
The synthesis of a new series of free-base, NiII and ZnII 2,3,12,13-tetra(ethynyl)-5,10,15,20-tetraphenyl porphyrins is described. Upon heating, two of the four ethynyl moieties undergo Bergman cyclization to afford the monocyclized 2,3-diethynyl-5,20-diphenylpiceno[10,11,12,13,14,15-jklmn]porphyrin in 30 %, 10 %, and trace yields, respectively. The structures of all products were investigated by using quantum chemical calculations and the free-base analogue was isolated and crystallized; all compounds show significant deviation from the idealized planar structure. No fully-cyclized bispiceno[20,1,2,3,4,5,10,11,12,13,14,15-fghij]porphyrin was isolated from the reaction mixture. To understand why only two of the four enthynyl groups undergo Bergman cyclization, the reaction coordinates were examined by using DFT at the PWPW91/cc-pVTZ(-f) level coupled to a continuum solvation model. The barrier to cyclization of the second pair of ethynyl groups was found to be 5.5 kcal mol−1 higher than the first, suggesting a negative cooperative effect and significantly slower rate for the second cyclization. Cyclization reactions for model porphyrin–enediynes with ethene- and H-functionality substitutions at the meso-phenyl rings were also examined, and found to have a similar barrier to diradical formation for the second cyclization event as for the first in these highly planar molecules. By enforcing an artificial 30° cant in two of the pyrrole rings of the porphyrin, the second barrier was increased by 2 kcal mol−1 in the ethene model system; this suggests that the disruption of the π conjugation of the extended porphyrin structure is the cause of the increased barrier to the second cyclization event.
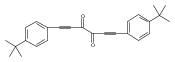
Co-reporter:Brigitte R. Spencer, Brian J. Kraft, Chris G. Hughes, Maren Pink, and Jeffrey M. Zaleski
Inorganic Chemistry 2010 Volume 49(Issue 24) pp:11333-11345
Publication Date(Web):November 23, 2010
DOI:10.1021/ic1011617
The spectroscopic, electronic, and DNA-binding characteristics of two novel ruthenium complexes based on the dialkynyl ligands 2,3-bis(phenylethynyl)-1,4,8,9-tetraaza-triphenylene (bptt, 1) and 2,3-bis(4-tert-butyl-phenylethynyl)-1,4,8,9-tetraaza-triphenylene (tbptt, 2) have been investigated. Electronic structure calculations of bptt reveal that the frontier molecular orbitals are localized on the pyrazine-dialkynyl portion of the free ligand, a property that is reflected in a red shift of the lowest energy electronic transition (1: λmax = 393 nm) upon substitution at the terminal phenyl groups (2: λmax = 398 nm). Upon coordination to ruthenium, the low-energy ligand-centered transitions of 1 and 2 are retained, and metal-to-ligand charge transfer transitions (MLCT) centered at λmax = 450 nm are observed for [Ru(phen)2bptt]2+(3) and [Ru(phen)2tbptt]2+(4). The photophysical characteristics of 3 and 4 in ethanol closely parallel those observed for [Ru(bpy)3]2+ and [Ru(phen)3]2+, indicating that the MLCT excited state is primarily localized within the [Ru(phen)3]2+ manifold of 3 and 4, and is only sparingly affected by the extended conjugation of the bptt framework. In an aqueous environment, 3 and 4 possess notably small luminescence quantum yields (3: ϕH2O = 0.005, 4: ϕH2O = 0.011) and biexponential decay kinetics (3: τ1 = 40 ns, τ2 = 230 ns; 4: τ1 ∼ 26 ns, τ2 = 150 ns). Addition of CT-DNA to an aqueous solution of 3 causes a significant increase in the luminescence quantum yield (ϕDNA = 0.045), while the quantum yield of 4 is relatively unaffected (ϕDNA = 0.013). The differential behavior demonstrates that tert-butyl substitution on the terminal phenyl groups inhibits the ability of 4 to intercalate with DNA. Such changes in intrinsic luminescence demonstrate that 3 binds to DNA via intercalation (Kb = 3.3 × 104 M−1). The origin of this light switch behavior involves two competing 3MLCT states similar to that of the extensively studied light switch molecule [Ru(phen)2dppz]2+. The solvent- and temperature-dependence of the luminescence of 3 reveal that the extended ligand aromaticity lowers the energy of the 3ππ* excited state into competition with the emitting 3MLCT state. Interconversion between these two states plays a significant role in the observed photophysics and is responsible for the dual emission in aqueous environments.
Co-reporter:Aurora E. Clark, Sibaprasad Bhattacharryya and Jeffrey M. Zaleski
Inorganic Chemistry 2009 Volume 48(Issue 9) pp:3926-3933
Publication Date(Web):August 30, 2008
DOI:10.1021/ic801117m
Density functional theory (DFT) has been used to study electronic perturbations induced by ancillary halogen ligation within metalloenediyne constructs, and the subsequent affect upon thermal Bergman cyclization temperatures. To isolate electronic from geometric components of Bergman cyclization thermodynamics, model diamine- and diphosphine-enediynes (L = 1,6-diamino- or 1,6-diphosphino-cis-1,5-hexadiyne-3-ene) of Mn(II), Cu(II), Zn(II), and Pd(II) with ancillary chloride ligands have been examined computationally and compared to more complex ethylenediamine-based metalloenediyne frameworks of the form MLX2 (X = Cl, Br, I; L = 1,4-dibenzyl-1,4-diaza-cyclododec-8-ene-6,10-diyne) with distorted square-planar (Cu(II)), Td (Zn(II)), and D4h (Pd(II)) geometries. In the latter systems, the ethylenediamine linkage restricts the conformation of the enediyne backbone, causing the alkyne termini separation to be nearly independent of metal geometry (3.75−3.82 Å). Within the Zn(II) family, steric effects are shown to induce conformational changes on the cyclization potential energy surface (PES) prior to the Bergman transition state, introducing distinct electron−electron repulsive interactions. Multiple metal and ligand conformations are also observed on the Cu(II) metalloenediyne cyclization PES. In contrast, square-planar Pd(II) compounds exhibit overlap between the out-of-plane halogen lone pairs and metal d orbitals, as well as the enediyne π system, reminiscent of an organometallic “push−pull” reaction mechanism. These systems have significantly higher predicted activation barriers toward cycloaromatization due to enhanced electron repulsion.
Co-reporter:Sibaprasad Bhattacharyya, Aurora E. Clark, Maren Pink and Jeffrey M. Zaleski
Inorganic Chemistry 2009 Volume 48(Issue 9) pp:3916-3925
Publication Date(Web):August 30, 2008
DOI:10.1021/ic801116q
The synthesis of novel metalloendiyne complexes MLRX2 (where L = 1,4-dibenzyl/diethyl-1,4-diaza-cyclododec-8-ene-6,10-diyne, X = halogen) are reported with their X-ray crystal structures and thermal Bergman cyclization temperatures. Two distinct types of constructs are obtained; the Zn(II) compounds are tetrahedral, while the Cu(II) and the Pd(II) compounds are all distorted- or square-planar. Each possesses structurally similar enediyne conformations and critical distances (3.75−3.88 Å). The tetragonal Cu(II) species all exhibit Bergman cyclization temperatures between 140 and 150 °C in the solid state, while the square-planar Pd(II) analogues possess similar critical distances but cyclize at significantly higher temperatures (205−220 °C). In contrast, the Zn(II) derivatives show a marked halogen dependence, with X = Cl having the highest Bergman cyclization temperature, which is comparable to the Pd(II) square-planar set, while the ZnLX2 compound with X = I shows the lowest Bergman cyclization temperature (144 °C), similar to the Cu(II) derivatives. Moreover, for the planar constructs, the R group has little influence on the cyclization temperatures; however, for the tetrahedral ZnLX2 compounds, the steric influence of the R group plays a more significant role in the cyclization reaction coordinate by influencing the stability of the precyclized intermediate. This complex set of results is best interpreted by a combination of steric contributions and electronic interactions between the halogen through space (in the case of Zn(II)) and through bonds (in the case of Pd(II)) and the π orbitals of the endiyne fragment. In contrast, for Cu(II) systems, the distorted square-planar geometry permits neither direct through space nor symmetry-allowed through bond communication between the orbital partners, and thus little variation in Bergman cyclization reactivity is observed.
Co-reporter:David F. Dye, Krishnan Raghavachari, Jeffrey M. Zaleski
Inorganica Chimica Acta 2008 Volume 361(Issue 4) pp:1177-1186
Publication Date(Web):3 March 2008
DOI:10.1016/j.ica.2007.10.049
The electronic and vibrational Raman spectra of octa-substituted (R = –SC10H21) Co- and Cu-porphyrazines are reported in their solid-state, mesophase, and isotropic liquid forms, as well as in THF solution. Their electronic spectra are composed of traditional Soret (CuS10 = 355 nm, CoS10 = 347 nm) and lower energy Q-bands (CuS10 = 669 nm, CoS10 = 639 nm), as well as a weaker, functionality-specific sulfur n → porphyrin π∗ feature (CuS10 = 500 nm; CoS10 = 447 nm). In contrast to the broad Q-band for CoS10 in all three neat phases, the lower energy analogue for CuS10 is markedly sharper in the microcrystalline state, but similarly broadens in the mesophase, indicative of long range macrocycle π–π interactions that persist even into the liquid state. The resonance (λ = 647 nm) and off-resonance (λ = 785 nm) Raman spectra of these materials in each phase exhibit four diagnostic vibrations; the Cα–Nm stretch (∼1540–1553) cm−1, Cβ–Cβ stretch (∼1450 cm−1), Cα–Cβ–Np stretch (∼1300–1315 cm−1), and Cα–Cβ stretch (∼1070 cm−1). For CoS10, these vibrations systematically shift to lower energy upon melting, while those for CuS10 collapse to degenerate sets. The differences in the electronic and vibrational profiles as a function of temperature suggest that the mesophase structure is governed by strong axial Co–S interactions for CoS10 which template macrocycle π–π stacking, while for CuS10 the same contacts exist, but they are phase dependent and markedly weaker. These inter-porphyrazine interactions are, therefore, responsible for the distinct differences in the melting and clearing temperatures of their respective mesophases. Finally, based on these diagnostic spectroscopic signatures, a photo-thermal, phase-switching mechanism is demonstrated with λ = 785 nm excitation at reduced temperatures, leading to the ability to spectrally monitor and phase change with a single photon source.Electronic and Raman spectroscopy of Co- and Cu-porphyrazine liquid crystals in all three phases reveal subtle, inter-macrocycle structural detail regarding metal–sulfur and π–π association within the layers. Photo-thermal laser excitation can disrupt this association and phase transition these materials, simultaneously monitoring their in situ state by Raman spectroscopy.
Co-reporter:Tillmann Köpke, Maren Pink and Jeffrey M. Zaleski
Chemical Communications 2006 (Issue 47) pp:4940-4942
Publication Date(Web):06 Oct 2006
DOI:10.1039/B611567E
Reaction of 2,3-dioxochlorins with benzeneselenic anhydride (BSA) results in the formation of unusual ring-contracted azetine derivatives that further react with BSA to afford porpholactones.
Co-reporter:Tillmann Köpke, Maren Pink and Jeffrey M. Zaleski
Organic & Biomolecular Chemistry 2006 vol. 4(Issue 22) pp:4059-4062
Publication Date(Web):04 Oct 2006
DOI:10.1039/B612776M
Photolysis of the Ni(II), Cu(II), and Zn(II) 2-diazo-3-oxochlorins generates 4-membered rings containing azeteoporphyrins.
Co-reporter:Sibaprasad Bhattacharyya, David F. Dye, Maren Pink and Jeffrey M. Zaleski
Chemical Communications 2005 (Issue 42) pp:5295-5297
Publication Date(Web):22 Sep 2005
DOI:10.1039/B509125J
Tetradentate metalloenediynes with strong imine and weaker thioether coordination serve as a geometrically non-rigid switch to drive thermal Bergman cyclization.
Co-reporter:Sibaprasad Bhattacharyya;Maren Pink;Mu-Hyun Baik
Angewandte Chemie 2005 Volume 117(Issue 4) pp:
Publication Date(Web):7 DEC 2004
DOI:10.1002/ange.200461825
Der Ring schließt sich schneller bei den MoIV-Komplexen von Endithiolat-Endiinen (siehe Schema; S gelb, Mo rot) als bei den entsprechenden nichtkomplexierten Endiinen. Erklärt wird diese Beobachtung mit einer langreichweitigen elektronischen Polarisation durch das Metallzentrum. Ligand-Metall-Charge-Transfer der C2S4-Einheit und Ladungsabstoßung im Übergangszustand senken die Aktivierungsbarriere der Cyclisierung.
Co-reporter:Sibaprasad Bhattacharyya;Maren Pink;Mu-Hyun Baik
Angewandte Chemie International Edition 2005 Volume 44(Issue 4) pp:
Publication Date(Web):7 DEC 2004
DOI:10.1002/anie.200461825
Dramatic differences are found in the temperatures at which Bergman cyclizations occur when MoIV–enedithiolate–enediyne complexes are used (see scheme; S: yellow, Mo: red), and this can be directly attributed to a long-range electronic polarization effect of the metal center. Ligand-to-metal charge transfer within the C2S4 substructure and differential charge repulsion in the transition state lead to a lowering of the barrier to cyclization.
Co-reporter:Aurora E. Clark, Ernest R. Davidson and Jeffrey M. Zaleski
Chemical Communications 2003 (Issue 23) pp:2876-2877
Publication Date(Web):22 Oct 2003
DOI:10.1039/B308633J
Time-dependent density functional theory shows that the photoreactivities of copper and zinc metalloenediynes derive from multi-configurational excited states involving the enediyne and pyridine π systems.
Co-reporter:Mahendra Nath, John C. Huffman and Jeffrey M. Zaleski
Chemical Communications 2003 (Issue 7) pp:858-859
Publication Date(Web):05 Mar 2003
DOI:10.1039/B212923J
The Bergman cyclization of simple diethynylporphyrinic-enediynes exhibits a double activation barrier to the formation of Bergman cyclized product. Addition of H-atom acceptor accelerates the formation of the picenoporphyrin, indicating that the second barrier is rate limiting.
Co-reporter:Elbert W. Schmitt, John C. Huffman and Jeffrey M. Zaleski
Chemical Communications 2001 (Issue 2) pp:167-168
Publication Date(Web):04 Jan 2001
DOI:10.1039/B008337M
Isostructural d10 metalloenediynes of
Cu(I), Pd(0) and Ag(I) exhibit
metal-dependent variations in their thermal Bergman cyclization
temperatures which correlate with differences in their alkyne termini
separations.
Co-reporter:Brian J. Kraft and Jeffrey M. Zaleski
New Journal of Chemistry 2001 vol. 25(Issue 10) pp:1281-1289
Publication Date(Web):11 Sep 2001
DOI:10.1039/B105693J
The
photochemical reactivities of 3-hydroxy-1,2,3-benzotriazine-4(3H)-one (1a) and tris[3-hydroxy-1,2,3-benzotriazine-4(3H)-one]iron(III) (1b) have been studied in solution and low temperature (5–77 K) glasses. Photoexcitation (λ345 nm) of 1a and 1b ultimately results in population of a ligand-centered excited state that releases N2. Electron
paramagnetic resonance reveals the presence of S
= 1 and S
= 0 diradical intermediates upon photolysis of 1a and 1b, respectively, at 4 K. These species convert to an S
= 1/2, nitrogen-centered monoradical species (aN
= 23 G) upon warming to 77 K ia H-atom abstraction from the matrix. In solution, the first intermediates observed
upon photolyses (λ
= 355 nm) of 1a and 1b are oxime ketenes (5: λ
= 385, 440 nm; 9: λ
= 390, 430, 740 nm) that are formed from collapse of the diradical to generate a 4-membered β-lactam ring. The decay kinetics for the oxime ketene 5 decay can be fitted to a biexponential expression representing a parallel reaction mechanism with an element of
reversibility. Thus, the data proposes the existence of an equilibrium between the oxime ketene and the spectroscopically
silent β-lactam intermediate, as well as a first or pseudo first-order solvent-dependent pathway for the oxime ketene. The kinetics for formation and decay of the ketene are strongly influenced by the presence of the Fe(III) center, which leads to an increase in the lifetime of the diradical in solution, and a retarded rate of formation for the oxime ketene 9. The solution lifetimes suggest that the diradical intermediates only persist sufficiently
long to react with bound solution substrates, whereas the ketene intermediates have suitable
kinetic viabilities to react bimolecularly in solution.
Co-reporter:Diwan S. Rawat and Jeffrey M. Zaleski
Chemical Communications 2000 (Issue 24) pp:2493-2494
Publication Date(Web):29 Nov 2000
DOI:10.1039/B007360L
The steric influences of the functional groups at the termini
of acyclic enediynes can dramatically affect the Bergman cyclization
temperatures of the resulting compounds.
Co-reporter:Tucker D. Maurer, Brian J. Kraft, Susan M. Lato, Andrew D. Ellington and Jeffrey M. Zaleski
Chemical Communications 2000 (Issue 1) pp:69-70
Publication Date(Web):06 Jan 2000
DOI:10.1039/A908005H
Visible wavelength ligand-to-metal (LMCT) activated
N2 release from
tris(3-hydroxy-1,2,3-benzotriazine-4(3H)-one]iron(III)
produces localized ligand radical intermediates capable of cleaving DNA and
represents a new chemical approach to photonuclease design for biological
applications.
Co-reporter:Meghan R. Porter, Akiko Kochi, Jonathan A. Karty, Mi Hee Lim and Jeffrey M. Zaleski
Chemical Science (2010-Present) 2015 - vol. 6(Issue 2) pp:NaN1026-1026
Publication Date(Web):2014/10/30
DOI:10.1039/C4SC01979B
Current approaches toward modulation of metal-induced Aβ aggregation pathways involve the development of small molecules that bind metal ions, such as Cu(II) and Zn(II), and interact with Aβ. For this effort, we present the enediyne-containing ligand (Z)-N,N′-bis[1-pyridin-2-yl-meth(E)-ylidene]oct-4-ene-2,6-diyne-1,8-diamine (PyED), which upon chelation of Cu(II) and Zn(II) undergoes Bergman-cyclization to yield diradical formation. The ability of this chelation-triggered diradical to modulate Aβ aggregation is evaluated relative to the non-radical generating control pyridine-2-ylmethyl-(2-{[(pyridine-2-ylmethylene)-amino]-methyl}-benzyl)-amine (PyBD). Variable-pH, ligand UV-vis titrations reveal pKa = 3.81(2) for PyBD, indicating it exists mainly in the neutral form at experimental pH. Lipinski's rule parameters and evaluation of blood–brain barrier (BBB) penetration potential by the PAMPA–BBB assay suggest that PyED may be CNS+ and penetrate the BBB. Both PyED and PyBD bind Zn(II) and Cu(II) as illustrated by bathochromic shifts of their UV-vis features. Speciation diagrams indicate that Cu(II)–PyBD is the major species at pH 6.6 with a nanomolar Kd, suggesting the ligand may be capable of interacting with Cu(II)–Aβ species. In the presence of Aβ40/42 under hyperthermic conditions (43 °C), the radical-generating PyED demonstrates markedly enhanced activity (2–24 h) toward the modulation of Aβ species as determined by gel electrophoresis. Correspondingly, transmission electron microscopy images of these samples show distinct morphological changes to the fibril structure that are most prominent for Cu(II)–Aβ cases. The loss of CO2 from the metal binding region of Aβ in MALDI-TOF mass spectra further suggests that metal–ligand–Aβ interaction with subsequent radical formation may play a role in the aggregation pathway modulation.
Co-reporter:Leigh J. K. Boerner, Maren Pink, Hyunsoo Park, Amanda LeSueur and Jeffrey M. Zaleski
Chemical Communications 2013 - vol. 49(Issue 21) pp:NaN2147-2147
Publication Date(Web):2013/01/29
DOI:10.1039/C3CC34471A
Thermal Bergman cyclization of Pt(II) dialkynylporphyrins reveals a marked reduction in the cyclization temperature relative to the free base and Zn(II) derivatives. In contrast, photogenerated 3ππ* population produces no detectable Bergman photocyclization, suggesting that the photoreactivities of the related free base and Zn(II) derivatives occurs via the 1ππ* state.
.jpg)