Co-reporter:Minying Cai; Udaya Kiran Marelli; Jennifer Bao; Johannes G. Beck; Florian Opperer; Florian Rechenmacher; Kaitlyn R. McLeod; Morgan R. Zingsheim; Lucas Doedens; Horst Kessler;Victor J. Hruby
Journal of Medicinal Chemistry 2015 Volume 58(Issue 16) pp:6359-6367
Publication Date(Web):July 28, 2015
DOI:10.1021/acs.jmedchem.5b00102
Human melanocortin receptors (hMCRs) have been challenging targets to develop ligands that are explicitly selective for each of their subtypes. To modulate the conformational preferences of the melanocortin ligands and improve the biofunctional agonist/antagonist activities and selectivities, we have applied a backbone N-methylation approach on Ac-Nle-c[Asp-His-d-Nal(2′)-Arg-Trp-Lys]-NH2 (Ac-Nle4-c[Asp5,d-Nal(2′)7,Lys10]-NH2), a nonselective cyclic peptide antagonist at hMC3R and hMC4R and an agonist at hMC1R and hMC5R. Systematic N-methylated derivatives of Ac-Nle4-c[Asp5,d-Nal(2′)7,Lys10]-NH2, with all possible backbone N-methylation combinations, have been synthesized and examined for their binding and functional activities toward melanocortin receptor subtypes 1, 3, 4, and 5 (hMCRs). Several N-methylated analogues are selective and potent agonists or antagonists for hMC1R or hMC5R or have selective antagonist activity for hMC3R. The selective hMC1R ligands show strong binding for human melanoma cells. We have also discovered the first universal antagonist (compound 19) for all subtypes of hMCRs.
Co-reporter:Yeon Sun Lee ; Vinod Kulkarani ; Scott M. Cowell ; Shou-wu Ma ; Peg Davis ; Katherine E. Hanlon ; Todd W. Vanderah ; Josephine Lai ; Frank Porreca ; Ruben Vardanyan ;Victor J. Hruby
Journal of Medicinal Chemistry 2011 Volume 54(Issue 1) pp:382-386
Publication Date(Web):December 3, 2010
DOI:10.1021/jm100982d
An SAR study on the Dmt-substituted enkephalin-like tetrapeptide with a N-phenyl-N-piperidin-4-ylpropionamide moiety at the C-terminal was performed and has resulted in highly potent ligands at μ and δ opioid receptors. In general, ligands with the substitution of d-Nle2 and halogenation of the aromatic ring of Phe4 showed highly increased opioid activities. Ligand 6 with good biological activities in vitro demonstrated potent in vivo antihyperalgesic and antiallodynic effects in the tail-flick assay.
Co-reporter:Domenica Torino ; Adriano Mollica ; Francesco Pinnen ; Federica Feliciani ; Gino Lucente ; Giancarlo Fabrizi ; Gustavo Portalone ; Peg Davis ; Josephine Lai ; Shou-Wu Ma ; Frank Porreca ;Victor J. Hruby
Journal of Medicinal Chemistry 2010 Volume 53(Issue 11) pp:4550-4554
Publication Date(Web):May 17, 2010
DOI:10.1021/jm1001343
New endomorphin-2 (EM-2) analogues incorporating (Z)-α,β-didehydrophenylalanine (ΔZPhe) in place of the native phenylalanine in EM-2 are reported. Tyr-Pro-ΔZPhe-Phe-NH2 {[ΔZPhe3]EM-2} (1), Tyr-Pro-Phe-ΔZPhe-NH2 {[ΔZPhe4]EM-2} (2), and Tyr-Pro-ΔZPhe-ΔZPhe-NH2 {[ΔZPhe3,4]EM-2}(3) have been synthesized, their opioid receptor binding affinities and tissue bioassay activities were determined, and their conformational properties were examined. Compound 2 shows high μ opioid receptor selectivity and μ agonist activity comparable to those of the native peptide. The conformation adopted in solution and in the crystal by N-Boc-Tyr-Pro-ΔZPhe-Phe-NH2 (8) is reported.
Co-reporter:Zhihua Liu, Hongchang Qu, Xuyuan Gu, Kwang-Soo Lee, Bryan Grossman, Vlad K. Kumirov, Victor J. Hruby
Tetrahedron Letters 2010 Volume 51(Issue 27) pp:3518-3520
Publication Date(Web):7 July 2010
DOI:10.1016/j.tetlet.2010.04.102
A significantly improved thio–Claisen rearrangement method was developed for preparing anti-β-functionalized γ,δ-unsaturated amino acids, which are extremely useful nonproteinogenic amino acids used in chemistry and biology research. The mild reaction condition successfully introduced base labile functional groups into the amino acids with excellent anti/syn selectivities.A significantly improved thio–Claisen rearrangement method was developed for preparing anti-β-functionalized γ,δ-unsaturated amino acids, which are extremely useful nonproteinogenic amino acids used in chemistry and biology research. The mild reaction condition successfully introduced base labile functional groups into the amino acids with excellent anti/syn selectivities.
Co-reporter:Yeon Sun Lee, Steve Fernandes, Vinod Kulkarani, Alexander Mayorov, Peg Davis, Shou-wu Ma, Kathy Brown, Robert J. Gillies, Josephine Lai, Frank Porreca, Victor J. Hruby
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 14) pp:4080-4084
Publication Date(Web):15 July 2010
DOI:10.1016/j.bmcl.2010.05.078
It has been known that co-administration of morphine with either cholecystokinin (CCK) receptor or melanocortin (MC) receptor antagonists enhance morphine’s analgesic efficacy by reducing serious side effects such as tolerance and addiction.1, 2, 3 and 4 Considering these synergistic effects, we have designed trivalent ligands in which all three different pharmacophores for opioid, CCK, and MC receptors are combined in such a way as to conserve their own topographical pharmacophore structures. These ligands, excluding the cyclic compound, were synthesized by solid phase synthesis using Rink-amide resin under microwave assistance in very high yields. These trivalent ligands bind to their respective receptors well demonstrating that the topographical pharmacophore structures for the three receptors were retained for receptor binding. Ligand 10 was a lead compound to show the best biological activities at all three receptors.
Co-reporter:Channa R. De Silva, Josef Vagner, Ronald Lynch, Robert J. Gillies, Victor J. Hruby
Analytical Biochemistry 2010 Volume 398(Issue 1) pp:15-23
Publication Date(Web):1 March 2010
DOI:10.1016/j.ab.2009.10.031
Lanthanide-based luminescent ligand binding assays are superior to traditional radiolabel assays due to improving sensitivity and affordability in high-throughput screening while eliminating the use of radioactivity. Despite significant progress using lanthanide(III)-coordinated chelators such as diethylenetriaminepentaacetic acid (DTPA) derivatives, dissociation-enhanced lanthanide fluoroimmunoassays (DELFIAs) have not yet been successfully used with more stable chelators (e.g., tetraazacyclododecyltetraacetic acid [DOTA] derivatives) due to the incomplete release of lanthanide(III) ions from the complex. Here a modified and optimized DELFIA procedure incorporating an acid treatment protocol is introduced for use with Eu(III)–DOTA-labeled peptides. Complete release of Eu(III) ions from DOTA-labeled ligands was observed using hydrochloric acid (2.0 M) prior to the luminescent enhancement step. [Nle4,d-Phe7]-α-melanocyte-stimulating hormone (NDP-α-MSH) labeled with Eu(III)–DOTA was synthesized, and the binding affinity to cells overexpressing the human melanocortin-4 (hMC4) receptor was evaluated using the modified protocol. Binding data indicate that the Eu(III)–DOTA-linked peptide bound to these cells with an affinity similar to its DTPA analogue. The modified DELFIA procedure was further used to monitor the binding of an Eu(III)–DOTA-labeled heterobivalent peptide to the cells expressing both hMC4 and cholecystokinin-2 (CCK-2) receptors. The modified assay provides superior results and is appropriate for high-throughput screening of ligand libraries.
Co-reporter:Hongchang Qu ; Minying Cai ; Alexander V. Mayorov ; Paolo Grieco ; Morgan Zingsheim ; Dev Trivedi ;Victor J. Hruby
Journal of Medicinal Chemistry 2009 Volume 52(Issue 12) pp:3627-3635
Publication Date(Web):May 27, 2009
DOI:10.1021/jm801300c
A new series of melanotropin analogues with His or Arg residues in the core pharmacophores of MTII, SHU9119, and Ac-NDP-γ-MSH-NH2 replaced by Pro or trans-/cis-4-guanidinyl-Pro derivatives were designed and synthesized to introduce selectivity toward the human melanocortin 4 receptor (hMC4R). Analogues 1, 2, 3, 6, 7, 8 were found to be hMC4R selective. Second messenger studies have demonstrated that analogues 1 and 2 are insurmountable inhibitors of MTII agonist activity at the hMC4R. Molecular modeling studies suggest that the hMC4R selectivity is due to a β-turn shift induced by the Pro ring that makes the global minimum structures of these analogues resemble the NMR solution structure of the hASIP melanocortin receptor binding motif. Substitution of His in MTII also provided functional selectivity for the hMC3R or the hMC4R. These findings are important for a better understanding of the selectivity mechanism at the hMC3R/hMC4R and the development of therapeutic ligands selectively targeting the hMC4R.
Co-reporter:Josef Vagner, Hongchang Qu, Victor J Hruby
Current Opinion in Chemical Biology 2008 Volume 12(Issue 3) pp:292-296
Publication Date(Web):June 2008
DOI:10.1016/j.cbpa.2008.03.009
The demand for modified peptides with improved stability profiles and pharmacokinetic properties is driving extensive research effort in this field. Many structural modifications of peptides guided by rational design and molecular modeling have been established to develop novel synthetic approaches. Recent advances in the synthesis of conformationally restricted building blocks and peptide bond isosteres are discussed.
Co-reporter:Paolo Grieco ; Minying Cai ; Lu Liu ; Alexander Mayorov ; Kevin Chandler ; Dev Trivedi ; Guangxin Lin ; Pietro Campiglia ; Ettore Novellino ;Victor J. Hruby
Journal of Medicinal Chemistry 2008 Volume 51(Issue 9) pp:2701-2707
Publication Date(Web):April 15, 2008
DOI:10.1021/jm701181n
Differentiation of the physiological role of the melanocortin receptor 5 MC5R from that of other melanocortin receptors will require development of high affinity and selective antagonists. To date, a few synthetic antagonist ligands active at hMC5 receptor are available, but most do not have appreciable selectivity. With the aim to gain more potent and selective antagonists for the MC5R ligands, we have designed, synthesized, and pharmacologically characterized a series of alkylthioaryl-bridged macrocyclic peptide analogues derived from MT-II and SHU9119. These 20-membered macrocycles were synthesized by a tandem combination using solid phase peptide synthesis and microwave-assisted reactions. Biological assays for binding affinities and adenylate cyclase activities for the hMC1R, hMC3R, hMC4R, and hMC5R showed that three analogues, compounds, 9, 4, and 7, are selective antagonists at the hMC5 receptor. In particular, compound 9(PG-20N) is a selective and competitive hMC5R antagonist, with IC50 of 130 ± 11 nM, and a pA2 value of 8.3, and represents an important tool for further biological investigations of the hMC5R. Compounds 4 and 7 (PG14N, PG17N) show potent and selective allosteric inhibition at hMC5R with IC50 values of 38 ± 3 nM and 58 ± 6 nM, respectively. Compound 9 will be used to further investigate and more clearly understand the physiological roles played by the MC5 receptor in humans and other animals.
Co-reporter:Takashi Yamamoto ; Padma Nair ; Josef Vagner ; Tally Largent-Milnes ; Peg Davis ; Shou-wu Ma ; Edita Navratilova ; Sharif Moye ; Suneeta Tumati ; Josephine Lai ; Henry I. Yamamura ; Todd W. Vanderah ; Frank Porreca ;Victor J. Hruby
Journal of Medicinal Chemistry 2008 Volume 51(Issue 5) pp:1369-1376
Publication Date(Web):February 12, 2008
DOI:10.1021/jm070332f
A series of bifunctional peptides with opioid agonist and substance P antagonist bioactivities were designed with the concept of overlapping pharmacophores. In this concept, the bifunctional peptides were expected to interact with each receptor separately in the spinal dorsal horn where both the opioid receptors and the NK1 receptors were found to be expressed, to show an enhanced analgesic effect, no opioid-induced tolerance, and to provide better compliance than coadministration of two drugs. Compounds were synthesized using a two-step combinatorial method for C-terminal modification. In the method, the protected C-terminal-free carboxyl peptide, Boc-Tyr(tBu)-d-Ala-Gly Phe-Pro-Leu-Trp(Boc)-OH, was synthesized as a shared intermediate using Fmoc solid phase chemistry on a 2-chlorotrityl resin. This intermediate was esterified or amidated in solution phase. The structure–activity relationships (SAR) showed that the C-terminus acted as not only a critical pharmacophore for the substance P antagonist activities, but as an address region for the opioid agonist pharmacophore that is structurally distant from the C-terminal. Among the peptides, H-Tyr-d-Ala-Gly-Phe-Pro-Leu-Trp-NH-Bzl (3) demonstrated high binding affinities at both δ and μ receptors (Ki = 10 and 0.65 nM, respectively) with efficient agonist functional activity in the mouse isolated vas deferens (MVD) and guinea pig isolated ileum (GPI) assays (IC50 = 50 and 13 nM, respectively). Compound 3 also showed a good antagonist activity in the GPI assay with substance P stimulation (Ke = 26 nM) and good affinity for the hNK1 receptor (Ki = 14 nM). Consequently, compound 3 is expected to be a promising and novel type of analgesic with bifunctional activities.
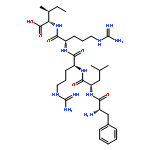
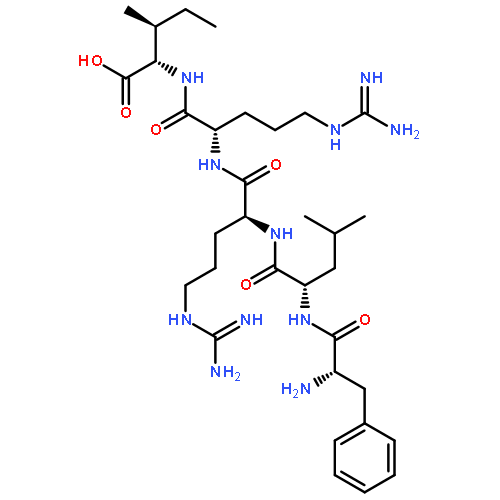
Co-reporter:Alexander V. Mayorov ; Minying Cai ; Erin S. Palmer ; Matthew M. Dedek ; James P. Cain ; April R. Van Scoy ; Bahar Tan ; Josef Vagner ; Dev Trivedi ;Victor J. Hruby
Journal of Medicinal Chemistry 2008 Volume 51(Issue 2) pp:187-195
Publication Date(Web):December 19, 2007
DOI:10.1021/jm070461w
A variety of dicarboxylic acid linkers introduced between the α-amino group of Pro6 and the ϵ-amino group of Lys10 of the cyclic lactam α-melanocyte-stimulating hormone (α-MSH)-derived Pro6-d-Phe7/d-Nal(2′)7-Arg8-Trp9-Lys10-NH2 pentapeptide template lead to nanomolar range and selective hMC3R agonists and antagonists. Replacement of the Pro6 residue and the dicarboxylic acid linker with 2,3-pyrazine-dicarboxylic acid furnished a highly selective nanomolar range hMC3R partial agonist (analogue 12, c[CO-2,3-pyrazine-CO-d-Phe-Arg-Trp-Lys]-NH2, EC50 = 27 nM, 70% max cAMP) and an hMC3R antagonist (analogue 13, c[CO-2,3-pyrazine-CO-d-Nal(2′)-Arg-Trp-Lys]-NH2, IC50 = 23 nM). Modeling experiments suggest that 2,3-pyrazinedicarboxylic acid stabilizes a β-turn-like structure with the d-Phe/d-Nal(2′) residues, which explains the high potency of the corresponding peptides. Placement of a Nle residue in position 6 produced a hMC3R/hMC5R antagonist (analogue 15, c[CO-(CH2)2-CO-Nle-d-Nal(2′)-Arg-Trp-Lys]-NH2, IC50 = 12 and 17 nM, respectively), similarly to the previously described cyclic γ-melanocyte-stimulating hormone (γ-MSH)-derived hMC3R/hMC5R antagonists. These newly developed melanotropins will serve as critical biochemical tools for elucidating the full spectrum of functions performed by the physiologically important melanocortin-3 receptor.
Co-reporter:Alexer V. Mayorov;So-Yeop Han;Minying Cai;Matthew R. Hammer;Dev Trivedi;Victor J. Hruby
Chemical Biology & Drug Design 2006 Volume 67(Issue 5) pp:
Publication Date(Web):1 JUN 2006
DOI:10.1111/j.1747-0285.2006.00383.x
The effects of the linker arm rigidity and size on melanocortin receptor selectivity were explored in a series of compounds using cyclic lactam α-melanocyte-stimulating hormone template. A variety of dicarboxylic acid linkers introduced between the α-amino group of His6 and the ɛ-amino group of Lys10 lead to high-affinity, selective human melanocortin receptor-1 and -5 (hMC1R and hMC5R) antagonists. The incorporation of hydrophilic functions into the linker arm was found to be unfavorable for both binding potency and receptor selectivity. Analogs 8 and 9 containing highly conformationally constrained hydrophobic linkers (m- and p-phthalic acids) were found to be selective nanomolar range hMC1R antagonists (IC50 = 7 and 4 nm, respectively), whereas the employment of a small conformationally constrained linker (maleic acid) resulted in a high-affinity (IC50 = 19 nm) and selective hMC5R antagonist (analog 12). These newly developed melanotropins will serve as critical biochemical tools for elucidating the full spectrum of functions performed by the physiologically important melanocortin-1 and -5 receptors.
Co-reporter:Minying Cai;Eva V. Varga;Magda Stankova;Alexer Mayorov;Joseph W. Perry;Henry I. Yamamura;Dev Trivedi;Victor J. Hruby
Chemical Biology & Drug Design 2006 Volume 68(Issue 4) pp:
Publication Date(Web):9 NOV 2006
DOI:10.1111/j.1747-0285.2006.00432.x
Melanocortin hormones and neurotransmitters regulate a vast array of physiologic processes by interacting with five G-protein-coupled melanocortin receptor types. In the present study, we have systematically studied the regulation of individual human melanocortin receptor wild subtypes using a synthetic rhodamine-labeled human melanotropin agonist and antagonist, arrestins fused to green fluorescent protein in conjunction with two-photon fluorescence laser scanning microscopy and confocal microscopy. Stimulation of the melanocortin receptors by its cognate agonist triggered rapid arrestin recruitment and receptor internalization for all four human melanocortin receptors examined. Antagonists-bound melanocortin receptors, on the other hand, did not recruit β-arrestins, and remained in the cell membrane even after long-term (30 min) treatment. Agonist-mediated internalization of all melanocortin receptor subtypes was sensitive to inhibitors of clathrin-dependent endocytosis, but not to caveolae inhibitors. In summary, agonist-mediated internalization of all subtypes of melanocortin receptors are dependent upon β-arrestin-mediated clathrin-coated pits, whereas, β-arrestin-2 conjugated green fluorescence protein (β-arrestin-2-GFP) recruitment is not dependent on protein kinase A activation. Real time two-photon fluorescence laser scanning microscopy is a most powerful tool to study the dynamic processes in living cells and tissues, without inflicting significant and often lethal damage to the specimen.
Co-reporter:Adriano Mollica, Peg Davis, Shou-Wu Ma, Frank Porreca, Josephine Lai, Victor J. Hruby
Bioorganic & Medicinal Chemistry Letters 2006 Volume 16(Issue 2) pp:367-372
Publication Date(Web):15 January 2006
DOI:10.1016/j.bmcl.2005.09.080
Biphalin is a linear octapeptide with strong opioid activity. Its structure is based on two identical sequences derived from enkephalins joined C-terminal to C-terminal by an hydrazide bridge (Tyr-D-Ala-Gly-Phe-NH-NH ← Phe ← Gly ← D-Ala ← Tyr). In this study we present the design, synthesis, and biological evaluation of the first cyclic biphalin analogues. d-Alanine residues in positions 2, 2′ of the parent peptide were replaced by d- and l-cysteine and an intramolecular disulfide bond between the cysteine thiol groups was introduced. We obtained two cyclic analogues with quite different biological profiles.Design, synthesis, and biological evaluation of the first cyclic biphalin analogues are reported. d-Alanine residues in positions 2, 2′ of the parent peptide were replaced by d- and l-cysteine and an intramolecular disulfide bridge has been established. Two cyclic biphalin analogues, with quite different biological profiles, have been described.
Co-reporter:Ravil R. Petrov, Ruben S. Vardanyan, Yeon S. Lee, Shou-wu Ma, Peg Davis, Lucinda J. Begay, Josephine Y. Lai, Frank Porreca, Victor J. Hruby
Bioorganic & Medicinal Chemistry Letters 2006 Volume 16(Issue 18) pp:4946-4950
Publication Date(Web):15 September 2006
DOI:10.1016/j.bmcl.2006.06.040
An enkephalin analogue coupled to ‘aminofentanyl’ has been synthesized and tested for biological activities at the μ and δ opioid receptors. Aminofentanyl which represents a structural derivative of fentanyl has been synthesized by acylation of 1-(2-phenethyl)-4-(N-anilino)piperidine with phthaloyl protected β-alaninyl chloride in the presence of DIPEA, followed by deprotection with hydrazine hydrate. Aminofentanyl has also been successfully acylated with ethyl isocyanate, various acid anhydrides, to further investigate structure–activity relationships of these new fentanyl derivatives. Among the new derivatives compound 7 which carries a Tyr-d-Ala-Gly-Phe opioid message sequence showed good opioid affinity (1 nM at both δ and μ opioid receptors) and bioactivity (34.9 nM in MVD and 42 nM in GPI/LMMP bioassays).The synthesis and results of binding affinity and in vitro bioassays of novel fentanyl analogues are reported.
Co-reporter:Adriano Mollica, Peg Davis, Shou-Wu Ma, Josephine Lai, Frank Porreca, Victor J. Hruby
Bioorganic & Medicinal Chemistry Letters 2005 Volume 15(Issue 10) pp:2471-2475
Publication Date(Web):16 May 2005
DOI:10.1016/j.bmcl.2005.03.067
Biphalin is a potent opioid peptide agonist, with a palandromic structure, composed of two enkephalin-like active fragments connected tail to tail by a hydrazine linker (Tyr-D-Ala-Gly-Phe-NH-NH<-Phe<-Gly<-D-Ala<-Tyr). This study presents the synthesis and in vitro bioassays of six new biphalin analogues with three different non-hydrazine linkers, some of which have higher binding affinity and bioactivity than biphalin.This study presents the synthesis and in vitro bioassay of new biphalin analogues with three novel non-hydrazine linkers.
Co-reporter:Alexander V. Mayorov, Minying Cai, Erin S. Palmer, Zhihua Liu, James P. Cain, Josef Vagner, Dev Trivedi, Victor J. Hruby
Peptides (October 2010) Volume 31(Issue 10) pp:1894-1905
Publication Date(Web):1 October 2010
DOI:10.1016/j.peptides.2010.06.026
A novel hybrid melanocortin pharmacophore was designed based on the pharmacophores of the agouti-signaling protein (ASIP), an endogenous melanocortin antagonist, and α-melanocyte-stimulating hormone (α-MSH), an endogenous melanocortin agonist. The designed hybrid ASIP/MSH pharmacophore was explored in monomeric cyclic, and cyclodimeric templates. The monomeric cyclic disulfide series yielded peptides with hMC3R-selective non-competitive binding affinities. The direct on-resin peptide lactam cyclodimerization yielded nanomolar range (25–120 nM) hMC1R-selective full and partial agonists in the cyclodimeric lactam series which demonstrates an improvement over the previous attempts at hybridization of MSH and agouti protein sequences. The secondary structure-oriented pharmacophore hybridization strategy will prove useful in development of unique allosteric and orthosteric melanocortin receptor modulators. This report also illustrates the utility of peptide cyclodimerization for the development of novel GPCR peptide ligands.Graphical abstractDownload full-size imageResearch highlights▶ A novel ASIP/MSH hybrid pharmacophore has been designed. ▶ A direct on-resin peptide lactam cyclodimerization is reported. ▶ Cyclodimeric ASIP/MSH hybrid peptides displayed nM range hMC1R agonist activities.





![Propanamide, N-(3,4-difluorophenyl)-N-[1-(phenylmethyl)-4-piperidinyl]-](http://img.cochemist.com/ccimg/202900/202859-29-8.png)
![Propanamide, N-(3,4-difluorophenyl)-N-[1-(phenylmethyl)-4-piperidinyl]-](http://img.cochemist.com/ccimg/202900/202859-29-8_b.png)

















![Butanoic acid, 4-oxo-4-[phenyl[1-(2-phenylethyl)-4-piperidinyl]amino]-](http://img.cochemist.com/ccimg/53000/52994-23-7.png)
![Butanoic acid, 4-oxo-4-[phenyl[1-(2-phenylethyl)-4-piperidinyl]amino]-](http://img.cochemist.com/ccimg/53000/52994-23-7_b.png)








![Benzene, [(3-iodo-2-propynyl)oxy]-](http://img.cochemist.com/ccimg/1800/1725-81-1.png)
![Benzene, [(3-iodo-2-propynyl)oxy]-](http://img.cochemist.com/ccimg/1800/1725-81-1_b.png)



![N-phenyl-n-[1-(2-phenylethyl)piperidin-4-yl]propanamide](http://img.cochemist.com/ccimg/500/437-38-7.png)
![N-phenyl-n-[1-(2-phenylethyl)piperidin-4-yl]propanamide](http://img.cochemist.com/ccimg/500/437-38-7_b.png)

