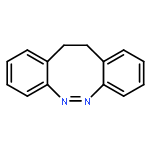
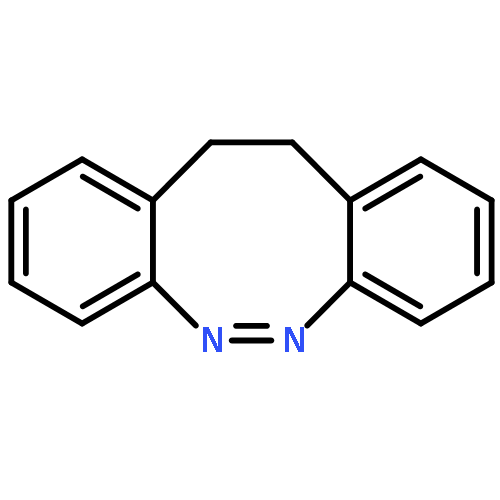
Co-reporter:Meihong Yang;Chunyan Huo;Anyang Li;Yibo Lei;Le Yu;Chaoyuan Zhu
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 19) pp:12185-12198
Publication Date(Web):2017/05/17
DOI:10.1039/C7CP00102A
The Zhu–Nakamura formulas based on on-the-fly trajectory surface hopping dynamics simulations at the two-state-averaged CASSCF level were employed to investigate the E → Z photoisomerization mechanisms of hemithioindigo–hemistilbene (HTI) upon S1 excitation. Seven conical intersections were observed along the isomerization pathways, which were composed of double bond torsion, benzene ring torsion, inversion and pyramidalization motions, and only three of them were found to play a role in the dynamics simulations started at S1E-HTI. The dominant isomerization pathway proceeds via central double bond torsion together with pyramidal and tilt motions to some extent (hop via CI5) and accounts for all the reactive trajectories. On the other hand, the two pathways that involve the conical zones lie in the vicinity of the E-form Franck–Condon region (CI7) and proceed along the combined central double bond and benzene ring torsion route (CI3/CI4) with generation of the E products. Within the 332 simulated trajectories, 66 hop to the ground state and only 19 switch to the Z product. The estimated quantum yield of 0.057 (19 in 332) agrees well with the reported experimental value of 0.053 ± 0.016. The excited-state lifetimes span a wide region from hundreds of femtoseconds to several picoseconds, depending on the time for vibrational relaxation and number of cycles for periodical mixed mode torsion.
Co-reporter:Yibo Lei, Huiyu Wu, Xiaolei Zheng, Gaohong Zhai, Chaoyuan Zhu
Journal of Photochemistry and Photobiology A: Chemistry 2016 Volume 317() pp:39-49
Publication Date(Web):15 February 2016
DOI:10.1016/j.jphotochem.2015.10.025
By a newly developed algorithm to compute global nonadiabatic switching probability only using electronic adiabatic potential energy surfaces and its gradients, we could able to perform on-the-fly trajectory surface hopping molecular dynamics at the SA-CASSCF(14, 8)/6-31G* quantum level to investigate the photo-induced 1,3-cyclohexadiene ring opening reaction from 1,3-cyclohexadiene (CHD) to 1,3,5-hexatriene (HT). We found four conical intersection zones within three singlet low-lying electronic states, in which 1B/2A and 1A/2A are well known to be the most important conical intersection zones to govern dynamics of ring opening reaction. The present simulation gained insight into nonadiabatic dynamics in which 516 sampling trajectories out of 600 can switch back and forth between two involved potential energy surfaces not only at 1B/2A, but also at 1A/2A. This insight is the key to influence quantum yields and time scales of the ring opening reaction. We estimated time constants as 46 fs, 82 fs, and 120 fs, respectively for crossing 1B/2A and 1A/2A, and excited-state lifetime in comparison with corresponding experimental values 56 fs, 80 fs and 130 fs. We estimated quantum yield 0.47/0.53 to HT/CHD in comparison to experiment values 0.30/0.70 in the gas-phase and 0.41/0.59 in the condense-phase. Moreover, the present simulation within 600 sampling trajectories indicates that there is no five membered ring forming in agreement with experiment results and with prediction from the previous wave-packet simulation as a rare case.
Co-reporter:Gao-Hong Zhai, Pei Yang, Shao-Mei Wu, Yi-Bo Lei, Yu-Sheng Dou
Chinese Chemical Letters 2014 Volume 25(Issue 5) pp:727-731
Publication Date(Web):May 2014
DOI:10.1016/j.cclet.2014.01.050
The photochromic ring-opening reaction of spiropyran (SP) has been investigated by a realistic semiclassical dynamics simulation, accompanied by SA3-CASSCF(12,10)/MS-CASPT2 potential energy curves (PECs) of S0–S2. The main simulation results show the dominate pathway corresponds to the ring-opening process of trans-SP to form the most stable merocyanine (MC) product. These findings provide more important complementarity for interpreting experimental observations.A realistic semiclassical dynamics simulation shows the ring-opening reaction process from the excited spiropyran (SP) to the trans–cis–cis merocyanine (MC-TCC) then to MC-CCC, and finally to the stable MC-TTC product.
Co-reporter:Meihong Yang, Chunyan Huo, Anyang Li, Yibo Lei, Le Yu and Chaoyuan Zhu
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 19) pp:NaN12198-12198
Publication Date(Web):2017/04/12
DOI:10.1039/C7CP00102A
The Zhu–Nakamura formulas based on on-the-fly trajectory surface hopping dynamics simulations at the two-state-averaged CASSCF level were employed to investigate the E → Z photoisomerization mechanisms of hemithioindigo–hemistilbene (HTI) upon S1 excitation. Seven conical intersections were observed along the isomerization pathways, which were composed of double bond torsion, benzene ring torsion, inversion and pyramidalization motions, and only three of them were found to play a role in the dynamics simulations started at S1E-HTI. The dominant isomerization pathway proceeds via central double bond torsion together with pyramidal and tilt motions to some extent (hop via CI5) and accounts for all the reactive trajectories. On the other hand, the two pathways that involve the conical zones lie in the vicinity of the E-form Franck–Condon region (CI7) and proceed along the combined central double bond and benzene ring torsion route (CI3/CI4) with generation of the E products. Within the 332 simulated trajectories, 66 hop to the ground state and only 19 switch to the Z product. The estimated quantum yield of 0.057 (19 in 332) agrees well with the reported experimental value of 0.053 ± 0.016. The excited-state lifetimes span a wide region from hundreds of femtoseconds to several picoseconds, depending on the time for vibrational relaxation and number of cycles for periodical mixed mode torsion.