Co-reporter:Hongbo Zheng; Yiwen Dong; Lin Li; Bin Sun; Lei Liu; Huiqing Yuan;Hongxiang Lou
Journal of Medicinal Chemistry 2016 Volume 59(Issue 10) pp:5063-5076
Publication Date(Web):April 14, 2016
DOI:10.1021/acs.jmedchem.6b00484
Paraptosis is nonapoptotic cell death characterized by massive endoplasmic reticulum (ER)- or mitochondria-derived vacuoles. Induction of paraptosis offers significant advantages for the treatment of chemotherapy-resistant tumors compared with anticancer drugs that rely on apoptosis. Because some natural alkaloids induce paraptotic cell death, a novel series of benzo[a]quinolizidine derivatives were synthesized, and their antiproliferative activity and ability to induce cytoplasmic vacuolation were analyzed. Structural optimization led to the identification of the potent compound 22b, which inhibited cancer cell proliferation in vitro and in vivo and profoundly facilitated paraptosis-like cell death and induced caspase-dependent apoptosis. Further investigation revealed that 22b-mediated vacuolation originated from persistent ER stress and upregulation of LC3B. Paraptosis induced by benzo[a]quinolizidine derivatives thus represents an alternative strategy for cancer chemotherapy.
Co-reporter:Hongxiang Lou;Dawei Wang;Huiqing Yuan;Xiaobin Li;Ming Wang;Wei Wang;Yongqing Liu
Cancer Chemotherapy and Pharmacology 2016 Volume 77( Issue 1) pp:63-75
Publication Date(Web):2016/01/01
DOI:10.1007/s00280-015-2915-4
Malformin A1 (MA1), a cyclopentapeptide isolated from fungal origin, has been identified to induce varieties of intriguing biological activities. Here, we reported the mode of mechanism underlying MA1-mediated cytotoxicity through induction of apoptosis, necrosis and autophagy in prostate cancer (PCa) cells.Human PCa cells PC3 and LNCaP were treated with MA1, and cell viability, apoptosis, necrosis, mitochondrial damage, oxidative stress and autophagy were analyzed, respectively. Pharmacological inhibitors, transient transfection of plasmids and siRNAs were then used to identify the roles of oxidative stress and autophagy in MA1-triggered cell death.In both PC3 and LNCaP cells, MA1 inhibited cell proliferation and triggered oxidative stress via the rapid accumulation of reactive oxygen species and a decrease in mitochondrial transmembrane potential. Mitochondrial damage by MA1 triggered caspase activation and intracellular ATP deletion, leading to apoptosis and necrosis, respectively. Meanwhile, MA1 activated autophagy as indicated by conversion of LC3BI to LC3BII and increased GFP-tagged LC3B punctate dots. Pharmacological inhibition of autophagy or knocking down LC3B attenuated MA1-mediated cell death. Excessive oxidative stress and decreased ATP stimulated AMPK/mTOR pathway, which led to induction of MA1-mediated autophagy.Coaction of apoptotic, necrotic and autophagic cell death induced by mitochondrial damage defines a novel mechanism contributing to the growth suppression of MA1 in prostate cancer cells, and activation of autophagy might be a potential strategy for improving its chemotherapeutic effects.
Co-reporter:J Sun;C Y F Young;H Jiang;J Wei;Q Xu;Y Liu;H Yuan;H Lou
Cell Death & Disease 2013 Volume 4(Issue 8) pp:e761
Publication Date(Web):2013-08-01
DOI:10.1038/cddis.2013.285
We previously reported that marchantin M (Mar) is an active agent to induce apoptosis in human prostate cancer (PCa), but the molecular mechanisms of action remain largely unknown. Here, we demonstrate that Mar potently inhibited chymotrypsin-like and peptidyl-glutamyl peptide-hydrolyzing activities of 20S proteasome both in in vitro and intracellular systems and significantly induced the accumulation of polyubiquitinated proteins in PCa cells. The computational modeling analysis suggested that Mar non-covalently bound to active sites of proteasome β5 and β1 subunits, resulting in a non-competitive inhibition. Proteasome inhibition by Mar subsequently resulted in endoplasmic reticulum (ER) stress, as evidenced by elevated glucose-regulated protein 78 and CHOP, increased phospho-eukaryotic translation initiation factor 2α (eIF2α), splicing of X-box-binding protein-1 and dilation of the ER. However, Mar-mediated cell death was not completely impaired by a pan inhibitor of caspases. Further studies revealed that the Mar-induced cell death was greatly associated with the activation of autophagy, as indicated by the significant induction of microtubule-associated protein-1 light chain-3 beta (LC3B) expression and conversion. Electron microscopic and green fluorescent protein-tagged LC3B analyses further demonstrated the ability of autophagy induction by Mar. Time kinetic studies revealed that Mar induced a rapid and highly sustained processing of LC3B in treated cells and simultaneously decreased the expression of p62/SQSTM1. Pharmacological blockade or knockdown of LC3B and Atg5 attenuated Mar-mediated cell death. The autophagic response triggered by Mar required the activation of RNA-dependent protein kinase-like ER kinase/eIF2α and suppression of the phosphatidylinositol-3 kinase/Akt/mammalian target of rapamycin axis via preventing activation and expression of Akt. Our results identified a novel mechanism for the cytotoxic effect of Mar, which strengthens it as a potential agent in cancer chemotherapy.
Co-reporter:Yan Ding;Xiaoling Wang;Aihui Xu;Xia Xu;Keli Tian;Charles Y.F. Young;Huiqing Yuan
Journal of Cellular Biochemistry 2011 Volume 112( Issue 3) pp:818-828
Publication Date(Web):
DOI:10.1002/jcb.22977
Abstract
We previously demonstrated that ectopic expression of neurotrophic peptide (NP) derived from saposin C promotes androgen receptor (AR) expression and transactivation in human prostate cancer cells. This prompted us to investigate how NP or saposin C can function in cells. We constructed plasmids expressing saposin C or a chimeric peptide of a viral TAT transduction domain and saposin C (TAT-saposin C) with His-tag. Intracellular localization of saposin C and NP was predominantly shown in transfected cells, while TAT-saposin C was detected around membrane and in cytosol by immunofluorescence staining. Furthermore, induction of the AR expression and activation of the AR transcriptional function were observed in cells transfected with saposin C or TAT-saposin C, compared to control cells transfected with an empty plasmid. The effects of saposin C and TAT-saposin C on AR activity were examined in the presence of inhibitors of GPCR, MAPK1/2, and PI3K/Akt. Interestingly, we found that these inhibitors only affect AR activities in cells with TAT-saposin C expression but not with saposin C expression. Immunostaining images showed that co-localization of saposin C, Src, and the AR occurred in transfected cells. Physical interactions of saposin C/NP, Src, and the AR were then demonstrated by co-immunoprecipitation assays. Blockage of Src activity by specific inhibitor led to a decrease in the saposin C-mediated enhancement of AR transactivity, suggesting that intracellular expression of saposin C caused stimulation of AR expression and activity by associations with Src in LNCaP cells. This effect may not be mediated by GPCR. J. Cell. Biochem. 112: 818–828, 2011. © 2010 Wiley-Liss, Inc.
Co-reporter:Guang-min Xi, Bin Sun, Hui-hui Jiang, Feng Kong, Hui-qing Yuan, Hong-xiang Lou
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 18) pp:6725-6733
Publication Date(Web):15 September 2010
DOI:10.1016/j.bmc.2010.07.055
P-glycoprotein (P-gp) is known to mediate multidrug resistance (MDR) by acting as an efflux pump to actively transport chemotherapeutic agents out of carcinoma cells. Inhibition of P-gp function may represent one of the strategies to reverse MDR. We have previously reported that marchantin C (MC), a macrocyclic bisbibenzyl compound from liverworts, exerts anti-tumor activity as an antimitotic agent. This study was designed to evaluate the possible modulatory effect of MC and its three synthetic derivatives (MC1, MC2 and MC3) on P-gp in VCR-resistant KB/VCR cells. Results of the cytotoxicity assay revealed that MC was the most potent inhibitor of cell proliferation in both KB and KB/VCR cells among these four compounds, while the three MC-derived chemicals had little anti-proliferative activity under the same condition. However, in P-gp-expressing MDR cells, analysis of potency of these compounds in enhancing cytotoxicity of VCR led to the identification of MC2 as a more effective chemical on reversal of resistance. Further study showed that MC2 was able to reduce efflux of rhodamine-123, and in turn, increase the accumulation of rhodamine-123 and adriamycin in KB/VCR cells, indicating that MC2 re-sensitized cells to VCR by inhibition of the P-gp transport activity. In addition, the combination of MC2 and VCR at a concentration that does not inhibit cell growth resulted in an induction of apoptosis in KB/VCR cells. These results suggest that MC2, as a novel and effective inhibitor of P-gp, may find potential application as an adjunctive agent with conventional chemotherapeutic drugs to reverse MDR in P-gp overexpressing cancer cells.
Co-reporter:Denglu Zhang, Yazhou Cui, Leilei Niu, Xia Xu, Keli Tian, Charles Y.F. Young, Hongxiang Lou, Huiqing Yuan
European Journal of Cell Biology (July 2014) Volume 93(Issue 7) pp:289-298
Publication Date(Web):1 July 2014
DOI:10.1016/j.ejcb.2014.05.004
Although several mechanisms behind resistance to docetaxel in castration-refractory prostate cancer (CRPC) have been investigated, molecular determinants of evolved resistance are still not entirely understood. Proteomics-based analysis in this study revealed that SOD2, associated with downregulation of reactive oxygen species (ROS), was significantly up-regulated in docetaxel-resistant (PC3/Doc) cells if compared to sensitive cells, and the expression of redox-regulated genes such as IGF-1R, CXCR4, and BCL2 was increased as well. Forced expression of SOD2 in sensitive cells led to the increase of IGF-1R and association with drug resistance, whereas silencing of SOD2 resulted in the decrease of IGF-1R at the protein level in resistant cells. Further study revealed that SOD2 acted as a negative regulator of β-arrestin1 that is an important adaptor responsible for degradation of IGF-1R via the changes in ROS, as evidenced by observations that an antioxidant agent substantially attenuated β-arrestin1 expression in vitro and in vivo. Finally, we found that blocking of IL6 that was up-regulated in resistant cells resulted in attenuation of SOD2 and STAT3, and simultaneously in increased expression of β-arrestin1. The modulation consequently led to the decreased IGF-1R at both protein and transcription levels. Together, our data provide a novel explanation that high level of IL6 stimulated SOD2 expression that, at least partially, contributed to the low level of ROS that would likely result in a sustained increase in the expression of IGF-1R through abolishment of β-arrestin1 in docetaxel resistant cells.
Co-reporter:Han Liu, Yi-qing Liu, Yong-qing Liu, Ai-hui Xu, Charles Y.F. Young, Hui-qing Yuan, Hong-xiang Lou
Chemico-Biological Interactions (5 December 2010) Volume 188(Issue 3) pp:598-606
Publication Date(Web):5 December 2010
DOI:10.1016/j.cbi.2010.07.024
Retigeric acid B (RB), a naturally occurring pentacyclic triterpenic acid, has been noted for its antifungal properties in vitro. Here, we observed that RB inhibited prostate cancer cell proliferation and induced cell death in a dose-dependent manner, but exerted very little inhibitory effect on noncancerous prostate epithelial cell viability. Treatment of androgen-independent PC-3 cells with RB caused a moderate increase in p21Cip1, and enforced the cell cycle arrest in the S phase. A block of S phase was accompanied with decreases in cyclin B, and increases in cyclin E and cyclin A proteins and phosphorylated retinoblastoma protein (pRb), whereas the expression of cdk2 remained almost unchanged in PC-3 cells exposed to RB. Moreover, RB significantly inhibited DNA synthesis with a dose-dependent reduction in the incorporation of BrdU into DNA, and enhanced apoptosis of PC-3 cells with induction of a higher ratio of Bax/Bcl-2 proteins, and activation of caspase-3 which, in turn, promoted the cleavage of poly (ADP-ribose) polymerase (PARP). However, pretreatment with the pan-caspase inhibitor z-VAD-fmk only partially alleviated RB-triggered apoptosis in PC-3 cells, suggesting the involvement of both caspase-dependent and caspase-independent pathways. Additionally, treatment of androgen-sensitive LNCaP cells with RB led to a reduction in the expression of androgen receptor (AR), and subsequently decreased the transactivity of AR. These observations help to support the search for promising candidates to treat prostate cancer.
Co-reporter:Leilei Niu, Jingti Deng, Fanghua Zhu, Nan Zhou, Keli Tian, Huiqing Yuan, Hongxiang Lou
Cancer Letters (28 June–1 July 2014) Volume 348(Issues 1–2) pp:126-134
Publication Date(Web):28 June 2014
DOI:10.1016/j.canlet.2014.03.019
As pro-inflammatory cytokines and chemokines contribute to the malignancy of many types of human cancer, we examined the anti-inflammatory effect of bisbibenzyls, a diverse bioactive group of naturally occurring compounds. Marchantin M (Mar M) was identified through a screening process of these compounds as a potent anti-inflammatory agent based on its capacity to inhibit LPS-induced IL6, IL1β and CCL2 expression in HUVECs and PBMCs without affecting cell proliferation. Since Mar M has been found to exhibit anticancer activity, we observed that Mar M treatment also resulted in decreases in the expressions of IL6, IL1β and TNFα in metastatic prostate cancer (PCa) cells. This effect was further confirmed in other cancer cell lines that express high level of pro-inflammatory cytokines. Furthermore, inactivation of NF-κB, a critical transcription factor controlling many pro-inflammatory cytokine expressions, was observed in Mar M-treated PCa cells as evidenced by decreased phosphor-p65 and subsequently phosphor-STAT3. Mar M also suppressed phosphorylation of IKBα, an inhibitor of NF-κB in the cytosol. However, reduced phosphor-p65 by Mar M was slightly increased when knockdown of IKBα, suggesting that Mar M may target upstream molecules of IKBα/NF-κB signaling. Finally, treatment with Mar M resulted in more enhanced-sensitivity of PCa cells to docetaxel-induced apoptosis than that of the IL6 blocking. Our study demonstrates the potential of the anti-inflammatory agent Mar M as an adjuvant to improve the efficacy of traditional anticancer agents such as docetaxel.

![Phenol, 2-methoxy-5-[[(tetrahydro-2H-pyran-2-yl)oxy]methyl]-](http://img.cochemist.com/ccimg/635000/634919-69-0.png)
![Phenol, 2-methoxy-5-[[(tetrahydro-2H-pyran-2-yl)oxy]methyl]-](http://img.cochemist.com/ccimg/635000/634919-69-0_b.png)






![Benzoic acid, 4-[5-(hydroxymethyl)-2-methoxyphenoxy]-, methyl ester](http://img.cochemist.com/ccimg/113300/113275-15-3.png)
![Benzoic acid, 4-[5-(hydroxymethyl)-2-methoxyphenoxy]-, methyl ester](http://img.cochemist.com/ccimg/113300/113275-15-3_b.png)
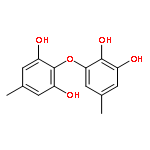
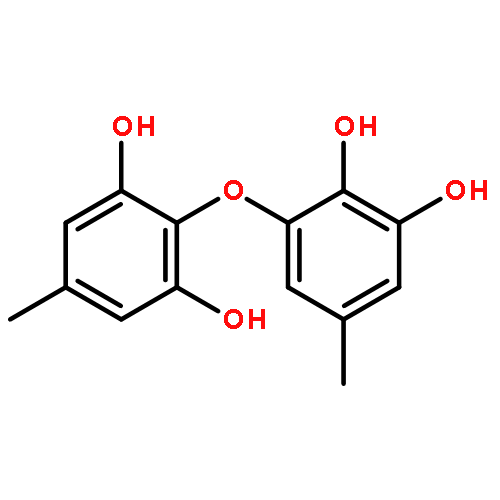
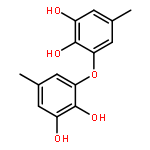
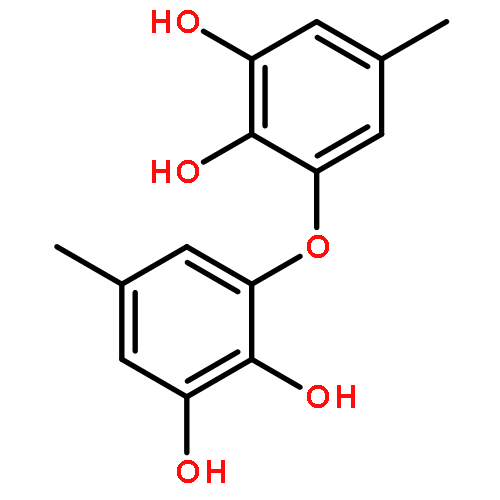


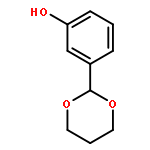
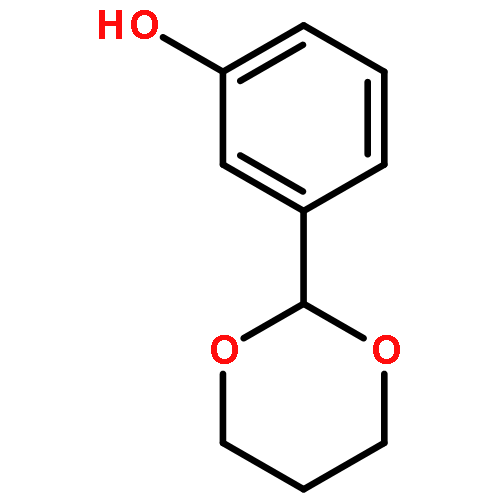




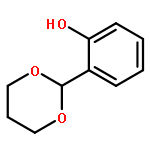
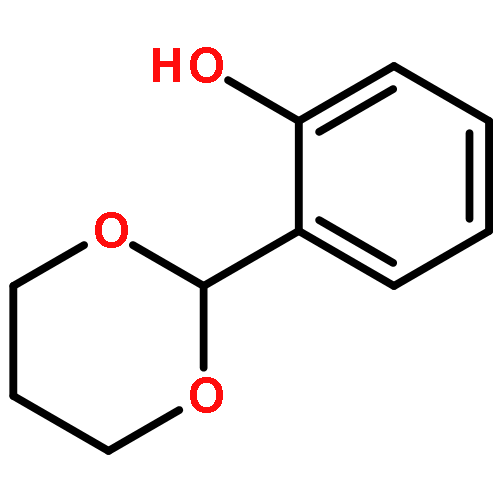


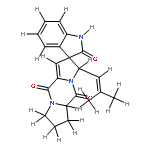
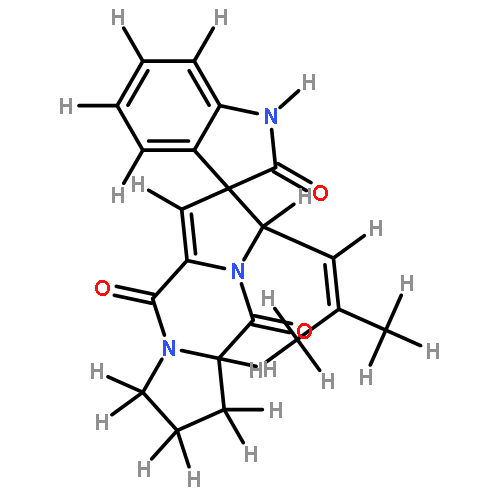
![2H,11H-Naphtho[2,1-b]pyrano[3,4-e]pyran-11-one,3,6-bis(acetyloxy)-4-[(acetyloxy)methyl]-1,3,4,4a,5,6,6a,12,12a,12b-decahydro-12-hydroxy-4,6a,12b-trimethyl-9-(3-pyridinyl)-,(3S,4R,4aR,6S,6aS,12R,12aS,12bS)-](http://img.cochemist.com/ccimg/147500/147444-03-9.png)
![2H,11H-Naphtho[2,1-b]pyrano[3,4-e]pyran-11-one,3,6-bis(acetyloxy)-4-[(acetyloxy)methyl]-1,3,4,4a,5,6,6a,12,12a,12b-decahydro-12-hydroxy-4,6a,12b-trimethyl-9-(3-pyridinyl)-,(3S,4R,4aR,6S,6aS,12R,12aS,12bS)-](http://img.cochemist.com/ccimg/147500/147444-03-9_b.png)
![5H,14H-Pyrrolo[1'',2'':4',5']pyrazino[1',2':1,6]pyrido[3,4-b]indole-5,14-dione,1,2,3,5a,6,11,12,14a-octahydro-9-methoxy-12-(2-methyl-1-propen-1-yl)-,(5aS,12S,14aS)-](http://img.cochemist.com/ccimg/119000/118974-02-0.png)
![5H,14H-Pyrrolo[1'',2'':4',5']pyrazino[1',2':1,6]pyrido[3,4-b]indole-5,14-dione,1,2,3,5a,6,11,12,14a-octahydro-9-methoxy-12-(2-methyl-1-propen-1-yl)-,(5aS,12S,14aS)-](http://img.cochemist.com/ccimg/119000/118974-02-0_b.png)
![(5aS,12S,14aS)-12-(2-methylprop-1-enyl)-1,2,3,5a,6,11,12,14a-octahydro-5H,14H-pyrrolo[1'',2'':4',5']pyrazino[2',1':6,1]pyrido[3,4-b]indole-5,14-dione](http://img.cochemist.com/ccimg/111800/111768-16-2.png)
![(5aS,12S,14aS)-12-(2-methylprop-1-enyl)-1,2,3,5a,6,11,12,14a-octahydro-5H,14H-pyrrolo[1'',2'':4',5']pyrazino[2',1':6,1]pyrido[3,4-b]indole-5,14-dione](http://img.cochemist.com/ccimg/111800/111768-16-2_b.png)
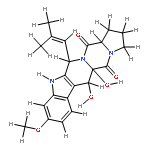
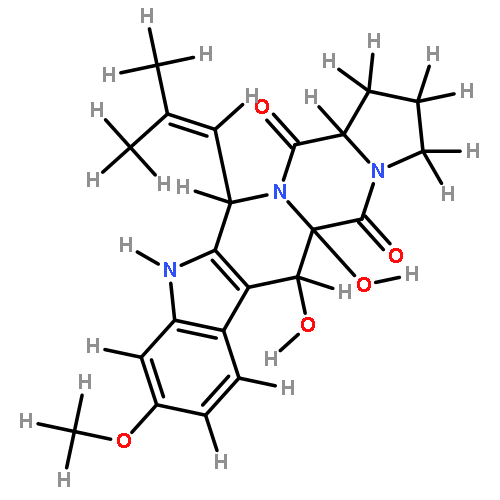
![1-Oxa-7-azaspiro[4.4]non-2-ene-4,6-dione,8-benzoyl-2-[(1S,2S,3Z)-1,2-dihydroxy-3-hexen-1-yl]-9-hydroxy-8-methoxy-3-methyl-,(5S,8S,9R)-](http://img.cochemist.com/ccimg/58600/58523-30-1.png)
![1-Oxa-7-azaspiro[4.4]non-2-ene-4,6-dione,8-benzoyl-2-[(1S,2S,3Z)-1,2-dihydroxy-3-hexen-1-yl]-9-hydroxy-8-methoxy-3-methyl-,(5S,8S,9R)-](http://img.cochemist.com/ccimg/58600/58523-30-1_b.png)
![(5aR,6S,12S,14aS)-5a,6-dihydroxy-12-(2-hydroxy-2-methylpropyl)-9-methoxy-1,2,3,5a,6,11,12,14a-octahydro-5H,14H-pyrrolo[1'',2'':4',5']pyrazino[1',2':1,6]pyrido[3,4-b]indole-5,14-dione](http://img.cochemist.com/ccimg/51200/51177-07-2.png)
![(5aR,6S,12S,14aS)-5a,6-dihydroxy-12-(2-hydroxy-2-methylpropyl)-9-methoxy-1,2,3,5a,6,11,12,14a-octahydro-5H,14H-pyrrolo[1'',2'':4',5']pyrazino[1',2':1,6]pyrido[3,4-b]indole-5,14-dione](http://img.cochemist.com/ccimg/51200/51177-07-2_b.png)
![(3S,8aS)-3-(1H-indol-3-ylmethyl)hexahydropyrrolo[1,2-a]pyrazine-1,4-dione](http://img.cochemist.com/ccimg/38200/38136-70-8.png)
![(3S,8aS)-3-(1H-indol-3-ylmethyl)hexahydropyrrolo[1,2-a]pyrazine-1,4-dione](http://img.cochemist.com/ccimg/38200/38136-70-8_b.png)

![methyl 4,6'-dimethoxy-6-methyl-3,4'-dioxo-3H-spiro[1-benzofuran-2,1'-cyclohexa[2,5]diene]-2'-carboxylate](http://img.cochemist.com/ccimg/2000/1900-29-4.png)
![methyl 4,6'-dimethoxy-6-methyl-3,4'-dioxo-3H-spiro[1-benzofuran-2,1'-cyclohexa[2,5]diene]-2'-carboxylate](http://img.cochemist.com/ccimg/2000/1900-29-4_b.png)