Co-reporter:Bryan J. Jones, Zsófia Bata, and Romas J. Kazlauskas
ACS Catalysis June 2, 2017 Volume 7(Issue 6) pp:4221-4221
Publication Date(Web):May 15, 2017
DOI:10.1021/acscatal.7b01108
Evolutionarily related hydroxynitrile lyases from rubber tree (HbHNL) and from Arabidopsis thaliana (AtHNL) follow different catalytic mechanisms with opposite enantioselectivity toward mandelonitrile. We hypothesized that the HbHNL-like mechanism evolved from an enzyme with an AtHNL-like mechanism. We created ancestor-like composite active sites in each scaffold to elucidate how this transition may have occurred. Surprisingly, a composite active site in HbHNL maintained (S)-selectivity, while the identical set of active site residues in AtHNL maintained (R)-selectivity. Composite active-site mutants that are (S)-selective without the Lys236 and Thr11 that are required for the classical (S)-HNL mechanism suggest a new mechanism. Modeling suggested a possibility for this new mechanism that does not exist in modern enzymes. Thus, the last common ancestor of HbHNL and AtHNL may have used an extinct mechanism, not the AtHNL-like mechanism. Multiple mechanisms are possible with the same catalytic residues and residues outside the active site strongly influence the mechanism and enantioselectivity.Keywords: ancestral enzyme; enantioselectivity; esterase; hydroxynitrile lyase; molecular dynamics; α/β-hydrolase fold;
Co-reporter:Hyeong Rae Lee, Hun Wook Lee, Youn-Woo Lee, Romas J. Kazlauskas, Tai Hyun Park
Biomass and Bioenergy 2017 Volume 105(Volume 105) pp:
Publication Date(Web):1 October 2017
DOI:10.1016/j.biombioe.2017.07.004
•Hot compressed water pretreatment of yellow poplar hydrolyzed xylan to xylose.•Subsequent peracetic acid treatment solubilized lignin leaving a glucan-rich solid.•Combined pretreatment removes more lignin than the sum of individual pretreatments.•Combined pretreatment could enhance enzymatic digestibility and reduce the amount of cellulase needed.•This fractionation of the major components of biomass allows separate uses for each component.Biomass forms a complex interwoven structure containing cellulose, hemicellulose and lignin that hinders enzymatic hydrolysis of cellulose. Enzymatic hydrolysis of the cellulose within yellow poplar (tulip tree) particles released only 9% of the total glucose in this study. To increase the accessibility of the cellulose component, wood particles were pretreated using hot compressed water and enzymatically-generated peracetic acid. The combined pretreatment started with hot compressed water (200 °C, 15 min), which selectively solubilized up to 90% of the xylan. The remaining solid was treated with peracetic acid (90 mM, 60 °C, 6 h), which solubilized up to 70% of the lignin. The remaining solid consisted of mainly glucan (∼75%) and corresponds to 87% of the glucan initially present in the yellow poplar particles. Hydrolysis of the remaining solid using a low loading of cellulase/β-glucosidase for 72 h released 90% of the glucose. The removal of the xylan and lignin structural barriers dramatically increased the cellulase accessibility to cellulose. The structural characteristics (crystallinity, functional group changes, morphology) of combined pretreated solid residue changed in a manner consistent with increased enzymatic digestibility. The combined pretreatment with hot compressed water and peracetic acid was more effective than either single pretreatment and more effective than the sum of the single pretreatments to remove xylan and lignin, thus demonstrating a cooperative effect of the two pretreatments. In addition, the combined pretreatment enhanced the accessibility of cellulases to the cellulose resulting in more efficient cellulose hydrolysis.
Co-reporter:Hyeong Rae Lee
Biotechnology and Bioprocess Engineering 2017 Volume 22( Issue 4) pp:405-412
Publication Date(Web):09 September 2017
DOI:10.1007/s12257-017-0139-7
Biomass contains cellulose, xylan and lignin in a complex interwoven structure that hinders enzymatic hydrolysis of the cellulose. To separate these components in yellow poplar biomass, we sequentially pretreated with dilute sulfuric acid and enzymatically-generated peracetic acid. In the first step, the dilute acid with microwave heating (140°C, 5 min) hydrolyzed 90% of xylan. The xylose yield in hydrolysate after dilute acid pretreatment was 83.1%. In the second step, peracetic acid (60°C, 6 h) removed up to 80% of lignin. This sequential pretreatment fractionated biomass into xylan and lignin, leaving a solid residue enriched in cellulose (~80%). The sequential pretreatment enhanced enzymatic digestibility of the cellulase by removal of the other components in biomass. The glucose yield after enzymatic hydrolysis was 90.5% at a low cellulase loading (5 FPU/g of glucan), which is 1.6 and 18 times higher than for dilute acid-pretreated biomass and raw biomass, respectively. This novel sequential pretreatment with dilute acid and peracetic acid efficiently separates the three major components of yellow poplar biomass, and reduces the amount of cellulase needed.
Co-reporter:Titu Devamani; Alissa M. Rauwerdink; Mark Lunzer; Bryan J. Jones; Joanna L. Mooney; Maxilmilien Alaric O. Tan; Zhi-Jun Zhang; Jian-He Xu; Antony M. Dean
Journal of the American Chemical Society 2016 Volume 138(Issue 3) pp:1046-1056
Publication Date(Web):January 6, 2016
DOI:10.1021/jacs.5b12209
Catalytic promiscuity is a useful, but accidental, enzyme property, so finding catalytically promiscuous enzymes in nature is inefficient. Some ancestral enzymes were branch points in the evolution of new enzymes and are hypothesized to have been promiscuous. To test the hypothesis that ancestral enzymes were more promiscuous than their modern descendants, we reconstructed ancestral enzymes at four branch points in the divergence hydroxynitrile lyases (HNL’s) from esterases ∼100 million years ago. Both enzyme types are α/β-hydrolase-fold enzymes and have the same catalytic triad, but differ in reaction type and mechanism. Esterases catalyze hydrolysis via an acyl enzyme intermediate, while lyases catalyze an elimination without an intermediate. Screening ancestral enzymes and their modern descendants with six esterase substrates and six lyase substrates found higher catalytic promiscuity among the ancestral enzymes (P < 0.01). Ancestral esterases were more likely to catalyze a lyase reaction than modern esterases, and the ancestral HNL was more likely to catalyze ester hydrolysis than modern HNL’s. One ancestral enzyme (HNL1) along the path from esterase to hydroxynitrile lyases was especially promiscuous and catalyzed both hydrolysis and lyase reactions with many substrates. A broader screen tested mechanistically related reactions that were not selected for by evolution: decarboxylation, Michael addition, γ-lactam hydrolysis and 1,5-diketone hydrolysis. The ancestral enzymes were more promiscuous than their modern descendants (P = 0.04). Thus, these reconstructed ancestral enzymes are catalytically promiscuous, but HNL1 is especially so.
Co-reporter:Hui Lin, Michael Travisano, and Romas J. Kazlauskas
ACS Chemical Biology 2016 Volume 11(Issue 9) pp:2568
Publication Date(Web):July 14, 2016
DOI:10.1021/acschembio.6b00376
In previous work, we evolved a population of Trichoderma citrinoviride in liquid cultures to speed up its asexual development cycle. The evolved population, called T-6, formed conidia 3 times sooner and in >1000-fold greater numbers. Here, we identify the steroid pregnenolone as a molecular signal for this different behavior. Media in which the ancestral T. citrinoviride population was grown (called ancestral spent media) contained a submerged conidiation inhibitor. Growing the evolved population T-6 in ancestral spent media eliminated the abundant formation of conidia. This inhibition depended on the amount and age of the ancestral spent medium and the time that the ancestral spent medium was added to the T-6 culture. Fractionation of the ancestral spent medium identified a hydrophobic inhibiting compound with a molecular weight less than 2000 g/mol. A combination of GC-MS, ELISA, and reaction with cholesterol oxidase identified it as pregnenolone. The addition of pregnenolone to cultures of T-6 inhibited submerged conidiation by inhibiting formation of conidiophores, while 10 other analogous steroids did not. Pregnenolone also inhibited submerged conidiation of Fusarium graminearum PH-1, a plant pathogen that causes head blight in wheat and barley. This identification of steroids as signal molecules in fungi creates opportunities to disrupt this signaling to control fungal behavior.
Co-reporter:Alissa Rauwerdink and Romas J. Kazlauskas
ACS Catalysis 2015 Volume 5(Issue 10) pp:6153
Publication Date(Web):September 9, 2015
DOI:10.1021/acscatal.5b01539
Enzymes within a family often catalyze different reactions. In some cases, this variety stems from different catalytic machinery, but in other cases, the machinery is identical; nevertheless, the enzymes catalyze different reactions. In this review, we examine the subset of α/β-hydrolase fold enzymes that contain the serine-histidine-aspartate catalytic triad. Despite having the same protein fold and the same core catalytic machinery, these enzymes catalyze 17 different reaction mechanisms. The most common reactions are hydrolysis of C–O, C–N, and C–C bonds (Enzyme Classification (EC) group 3), but other enzymes are oxidoreductases (EC group 1), acyl transferases (EC group 2), lyases (EC group 4), or isomerases (EC group 5). Hydrolysis reactions often follow the canonical esterase mechanism, but eight variations occur in which either the formation or cleavage of the acyl enzyme intermediate differs. The remaining eight mechanisms are lyase-type elimination reactions, which do not have an acyl enzyme intermediate and, in four cases, do not even require the catalytic serine. This diversity of mechanisms from the same catalytic triad stems from the ability of the enzymes to bind different substrates; from the requirements for different chemical steps imposed by these new substrates; and, only in about half of the cases, from additional hydrogen bond partners or additional general acids/bases in the active site. This detailed analysis shows that binding differences and noncatalytic residues create new mechanisms and are essential for understanding and designing efficient enzymes.Keywords: catalytic triad; divergent evolution; hydrolase; lyase; mechanism; oxyanion hole; X-ray structures
Co-reporter:Jun Huang, Bryan J. Jones, and Romas J. Kazlauskas
Biochemistry 2015 Volume 54(Issue 28) pp:4330-4341
Publication Date(Web):June 25, 2015
DOI:10.1021/acs.biochem.5b00333
α/β-Hydrolases are important enzymes for biocatalysis, but their stability often limits their application. We investigated a plant esterase, salicylic acid binding protein 2 (SABP2), as a model α/β-hydrolase. SABP2 shows typical stability to urea (unfolding free energy 6.9 ± 1.5 kcal/mol) and to heat inactivation (T1/215min 49.2 ± 0.5 °C). Denaturation in urea occurs in two steps, but heat inactivation occurs in a single step. The first unfolding step in urea eliminates catalytic activity. Surprisingly, we found that the first unfolding likely corresponds to the unfolding of the larger catalytic domain. Replacing selected amino acid residues with proline stabilized SABP2. Proline restricts the flexibility of the unfolded protein, thereby shifting the equilibrium toward the folded conformation. Seven locations for proline substitution were chosen either by amino acid sequence alignment with a more stable homologue or by targeting flexible regions in SABP2. Introducing proline in the catalytic domain stabilized SABP2 to the first unfolding in urea for three of five cases: L46P (+0.2 M urea), S70P (+0.1), and E215P (+0.9). Introducing proline in the cap domain did not stabilize SABP2 (two of two cases), supporting the assignment that the first unfolding corresponds to the catalytic domain. Proline substitutions in both domains stabilized SABP2 to heat inactivation: L46P (ΔT1/215min = +6.4 °C), S70P (+5.4), S115P (+1.8), S141P (+4.9), and E215P (+4.2). Combining substitutions did not further increase the stability to urea denaturation, but dramatically increased resistance to heat inactivation: L46P–S70P ΔT1/215min = +25.7 °C. This straightforward proline substitution approach may also stabilize other α/β-hydrolases.
Co-reporter:Heesung Eum;Hyun-Joon Ha
Advanced Synthesis & Catalysis 2014 Volume 356( Issue 17) pp:3585-3599
Publication Date(Web):
DOI:10.1002/adsc.201400510
Co-reporter:Dr. Jan von Langermann ;David M. Nedrud; Dr. Romas J. Kazlauskas
ChemBioChem 2014 Volume 15( Issue 13) pp:1931-1938
Publication Date(Web):
DOI:10.1002/cbic.201402081
Abstract
The natural substrate of hydroxynitrile lyase from rubber tree (HbHNL, Hevea brasiliensis) is acetone cyanohydrin, but synthetic applications usually involve aromatic cyanohydrins such as mandelonitrile. To increase the activity of HbHNL toward this unnatural substrate, we replaced active site residues in HbHNL with the corresponding ones from esterase SABP2 (salicylic acid binding protein 2). Although this enzyme catalyzes a different reaction (hydrolysis of esters), its natural substrate (methyl salicylate) contains an aromatic ring. Three of the eleven single-amino-acid-substitution variants of HbHNL reacted more rapidly with mandelonitrile. The best was HbHNL-L121Y, with a kcat 4.2 times higher and high enantioselectivity. Site-saturation mutagenesis at position 121 identified three other improved variants. We hypothesize that the smaller active site orients the aromatic substrate more productively.
Co-reporter:DeLu (Tyler) Yin;Vince M. Purpero;Ryota Fujii;Qing Jing
Chemistry - A European Journal 2013 Volume 19( Issue 9) pp:3037-3046
Publication Date(Web):
DOI:10.1002/chem.201202027
Abstract
Some serine hydrolases also catalyze a promiscuous reaction— reversible perhydrolysis of carboxylic acids to make peroxycarboxylic acids. Five X-ray crystal structures of these carboxylic acid perhydrolases show a proline in the oxyanion loop. Here, we test whether this proline is essential for high perhydrolysis activity using Pseudomonas fluorescens esterase (PFE). The L29P variant of this esterase catalyzes perhydrolysis 43-fold faster (kcat comparison) than the wild type. Surprisingly, saturation mutagenesis at the 29 position of PFE identified six other amino acid substitutions that increase perhydrolysis of acetic acid at least fourfold over the wild type. The best variant, L29I PFE, catalyzed perhydrolysis 83-times faster (kcat comparison) than wild-type PFE and twice as fast as L29P PFE. Despite the different amino acid in the oxyanion loop, L29I PFE shows a similar selectivity for hydrogen peroxide over water as L29P PFE (β0=170 vs. 160 M−1), and a similar fast formation of acetyl-enzyme (140 vs. 62 U mg−1). X-ray crystal structures of L29I PFE with and without bound acetate show an unusual mixture of two different oxyanion loop conformations. The type II β-turn conformation resembles the wild-type structure and is unlikely to increase perhydrolysis, but the type I β-turn conformation creates a binding site for a second acetate. Modeling suggests that a previously proposed mechanism for L29P PFE can be extended to include L29I PFE, so that an acetate accepts a hydrogen bond to promote faster formation of the acetyl-enzyme.
Co-reporter:Romas Kazlauskas
Chemistry & Biology 2012 Volume 19(Issue 4) pp:435-437
Publication Date(Web):20 April 2012
DOI:10.1016/j.chembiol.2012.04.004
A human enzyme variant, PON1-G3C9, accidentally catalyzes the hydrolysis of organophosphorus chemical weapons. In this issue of Chemistry & Biology, Goldsmith and coworkers describe a new PON1 variant with improved hydrolysis by several hundred fold; enough that it may protect animals from a toxic dose.
Co-reporter:DeLu (Tyler) Yin ; Romas J. Kazlauskas
Chemistry - A European Journal 2012 Volume 18( Issue 26) pp:8130-8139
Publication Date(Web):
DOI:10.1002/chem.201200052
Abstract
Several serine hydrolases catalyze a promiscuous reaction: perhydrolysis of carboxylic acids to form peroxycarboxylic acids. The working hypothesis is that perhydrolases are more selective than esterases for hydrogen peroxide over water. In this study, we tested this hypothesis, and focused on L29P-PFE (Pseudomonas fluorescens esterase), which catalyzes perhydrolysis of acetic acid 43-fold faster than wild-type PFE. This hypothesis predicts that L29P-PFE should be approximately 43-fold more selective for hydrogen peroxide than wild-type PFE, but experiments show that L29P-PFE is less selective. The ratio of hydrolysis to perhydrolysis of methyl acetate at different concentrations of hydrogen peroxide fit a kinetic model for nucleophile selectivity. L29P-PFE (β0=170 M−1) is approximately half as selective for hydrogen peroxide over water than wild-type PFE (β0=330 M−1), which contradicts the working hypothesis. An alternative hypothesis is that carboxylic acid perhydrolases increase perhydrolysis by forming the acyl-enzyme intermediate faster. Consistent with this hypothesis, the rate of acetyl-enzyme formation, measured by 18O-water exchange into acetic acid, was 25-fold faster with L29P-PFE than with wild-type PFE, which is similar to the 43-fold faster perhydrolysis with L29P-PFE. Molecular modeling of the first tetrahedral intermediate (Td1) suggests that a closer carbonyl group found in perhydrolases accepts a hydrogen bond from the leaving group water. This revised understanding can help design more efficient enzymes for perhydrolysis and shows how subtle changes can create new, unnatural functions in enzymes.
Co-reporter:Ian J. Colton;DeLu (Tyler) Yin;Pawel Grochulski
Advanced Synthesis & Catalysis 2011 Volume 353( Issue 13) pp:2529-2544
Publication Date(Web):
DOI:10.1002/adsc.201100459
Abstract
Lipase from Candida rugosa shows high enantioselectivity toward α-substituted chiral acids such as 2-arylpropionic acids. To understand how Candida rugosa lipase (CRL) distinguishes between enantiomers of chiral acids, we determined the X-ray crystal structure of a transition-state analog covalently linked to CRL. CRL shows moderate enantioselectivity (E=23) toward methyl 2-methoxy-2-phenylacetate, 1-methyl ester, favoring the (S)-enantiomer. We synthesized phosphonate (RC,RPSP)-3, which, upon reaction with CRL, mimics the transition state for hydrolysis of (S)-1-methyl ester, the fast-reacting enantiomer. An X-ray crystal structure of this complex shows a catalytically productive orientation with the phenyl ring in the hydrophobic tunnel of the lipase. Phe345 crowds the region near the substrate stereocenter. Computer modeling of the slow-reacting enantiomer examined four possible conformations for the corresponding slow-reacting enantiomer: three conformations where two substituents at the stereocenter have been exchanged relative to the fast-reacting enantiomer and one conformation with an umbrella-like inversion orientation. Each of these orientations disrupts the orientation of the catalytic histidine, but the molecular basis for disruption differs in each case showing that multiple mechanisms are required for high enantioselectivity.
Co-reporter:Dr. Yun Jiang;Dr. Krista L. Morley;Dr. Joseph D. Schrag; Dr. Romas J. Kazlauskas
ChemBioChem 2011 Volume 12( Issue 5) pp:768-776
Publication Date(Web):
DOI:10.1002/cbic.201000693
Abstract
Acyl transfer is a key reaction in biosynthesis, including synthesis of antibiotics and polyesters. Although researchers have long recognized the similar protein fold and catalytic machinery in acyltransferases and hydrolases, the molecular basis for the different reactivity has been a long-standing mystery. By comparison of X-ray structures, we identified a different oxyanion-loop orientation in the active site. In esterases/lipases a carbonyl oxygen points toward the active site, whereas in acyltransferases a NH of the main-chain amide points toward the active site. Amino acid sequence comparisons alone cannot identify such a difference in the main-chain orientation. To identify how this difference might change the reaction mechanism, we solved the X-ray crystal structure of Pseudomonas fluorescens esterase containing a sulfonate transition-state analogue bound to the active-site serine. This structure mimics the transition state for the attack of water on the acyl–enzyme and shows a bridging water molecule between the carbonyl oxygen mentioned above and the sulfonyl oxygen that mimics the attacking water. A possible mechanistic role for this bridging water molecule is to position and activate the attacking water molecule in hydrolases, but to deactivate the attacking water molecule in acyl transferases.
Co-reporter:Dr. Yun Jiang;Dr. Krista L. Morley;Dr. Joseph D. Schrag; Dr. Romas J. Kazlauskas
ChemBioChem 2011 Volume 12( Issue 5) pp:
Publication Date(Web):
DOI:10.1002/cbic.201190016
Co-reporter:Santosh Kumar Padhi, Ryota Fujii, Graig A. Legatt, Sara L. Fossum, Reto Berchtold, Romas J. Kazlauskas
Chemistry & Biology 2010 Volume 17(Issue 8) pp:863-871
Publication Date(Web):27 August 2010
DOI:10.1016/j.chembiol.2010.06.013
The α/β hydrolase superfamily contains mainly esterases, which catalyze hydrolysis, but also includes hydroxynitrile lyases, which catalyze addition of cyanide to aldehydes, a carbon-carbon bond formation. Here, we convert a plant esterase, SABP2, into a hydroxynitrile lyase using just two amino acid substitutions. Variant SABP2-G12T-M239K lost the ability to catalyze ester hydrolysis (<0.9 mU/mg) and gained the ability to catalyze the release of cyanide from mandelonitrile (20 mU/mg, kcat/KM = 70 min-1M-1). This variant also catalyzed the reverse reaction, formation of mandelonitrile with low enantioselectivity: 20% ee (S), E = 1.5. The specificity constant for the lysis of mandelontrile is 13,000-fold faster than the uncatalyzed reaction and only 1300-fold less efficient (kcat/KM) than hydroxynitrile lyase from rubber tree.Graphical AbstractFigure optionsDownload full-size imageDownload high-quality image (387 K)Download as PowerPoint slideHighlights► Esterases and hydroxynitrile lyases differ in their catalytic mechanisms ► Two amino acid replacement converts esterase to hydroxynitrile lyase
Co-reporter:De Lu (Tyler) Yin, Peter Bernhardt, Krista L. Morley, Yun Jiang, Jeremy D. Cheeseman, Vincent Purpero, Joseph D. Schrag and Romas J. Kazlauskas
Biochemistry 2010 Volume 49(Issue 9) pp:
Publication Date(Web):January 29, 2010
DOI:10.1021/bi9021268
Many serine hydrolases catalyze perhydrolysis, the reversible formation of peracids from carboxylic acids and hydrogen peroxide. Recently, we showed that a single amino acid substitution in the alcohol binding pocket, L29P, in Pseudomonas fluorescens (SIK WI) aryl esterase (PFE) increased the specificity constant of PFE for peracetic acid formation >100-fold [Bernhardt et al. (2005) Angew. Chem., Int. Ed. 44, 2742]. In this paper, we extend this work to address the three following questions. First, what is the molecular basis of the increase in perhydrolysis activity? We previously proposed that the L29P substitution creates a hydrogen bond between the enzyme and hydrogen peroxide in the transition state. Here we report two X-ray structures of L29P PFE that support this proposal. Both structures show a main chain carbonyl oxygen closer to the active site serine as expected. One structure further shows acetate in the active site in an orientation consistent with reaction by an acyl-enzyme mechanism. We also detected an acyl-enzyme intermediate in the hydrolysis of ε-caprolactone by mass spectrometry. Second, can we further increase perhydrolysis activity? We discovered that the reverse reaction, hydrolysis of peracetic acid to acetic acid and hydrogen peroxide, occurs at nearly the diffusion limited rate. Since the reverse reaction cannot increase further, neither can the forward reaction. Consistent with this prediction, two variants with additional amino acid substitutions showed 2-fold higher kcat, but Km also increased so the specificity constant, kcat/Km, remained similar. Third, how does the L29P substitution change the esterase activity? Ester hydrolysis decreased for most esters (75-fold for ethyl acetate) but not for methyl esters. In contrast, L29P PFE catalyzed hydrolysis of ε-caprolactone five times more efficiently than wild-type PFE. Molecular modeling suggests that moving the carbonyl group closer to the active site blocks access for larger alcohol moieties but binds ε-caprolactone more tightly. These results are consistent with the natural function of perhydrolases being either hydrolysis of peroxycarboxylic acids or hydrolysis of lactones.
Co-reporter:Shona Duncan;Qing Jing;Adrian Katona
Applied Biochemistry and Biotechnology 2010 Volume 160( Issue 6) pp:1637-1652
Publication Date(Web):2010 March
DOI:10.1007/s12010-009-8639-3
The recalcitrance of lignocellulosic biomass to enzymatic release of sugars (saccharification) currently limits its use as feedstock for biofuels. Enzymatic hydrolysis of untreated aspen wood releases only 21.8% of the available sugars due primarily to the lignin barrier. Nature uses oxidative enzymes to selectively degrade lignin in lignocellulosic biomass, but thus far, natural enzymes have been too slow for industrial use. In this study, oxidative pretreatment with commercial peracetic acid (470 mM) removed 40% of the lignin (from 19.9 to 12.0 wt.% lignin) from aspen and enhanced the sugar yields in subsequent enzymatic hydrolysis to about 90%. Increasing the amount of lignin removed correlated with increasing yields of sugar release. Unfortunately, peracetic acid is expensive, and concentrated forms can be hazardous. To reduce costs and hazards associated with using commercial peracetic acid, we used a hydrolase to catalyze the perhydrolysis of ethyl acetate generating 60–70 mM peracetic acid in situ as a pretreatment to remove lignin from aspen wood. A single pretreatment was insufficient, but multiple cycles (up to eight) removed up to 61.7% of the lignin enabling release of >90% of the sugars during saccharification. This value corresponds to a predicted 581 g of fermentable sugars from 1 kg of aspen wood. Improvements in the enzyme stability are needed before the enzymatically generated peracetic acid is a commercially viable alternative.
Co-reporter:Johnathan Gorke;Friedrich Srienc
Biotechnology and Bioprocess Engineering 2010 Volume 15( Issue 1) pp:40-53
Publication Date(Web):2010 February
DOI:10.1007/s12257-009-3079-z
Ionic liquids, also called molten salts, are mixtures of cations and anions that melt below 100°C. Typical ionic liquids are dialkylimidazolium cations with weakly coordinating anions such as (MeOSO3) or (PF6). Advanced ionic liquids such as choline citrate have biodegradable, less expensive, and less toxic anions and cations. Deep eutectic solvents are also included in the advanced ionic liquids. Deep eutectic solvents are mixtures of salts such as choline chloride and uncharged hydrogen bond donors such as urea, oxalic acid, or glycerol. For example, a mixture of choline chloride and urea in 1:2 molar ratio liquefies to form a deep eutectic solvent. Their properties are similar to those of ionic liquids. Water-miscible ionic liquids as cosolvents with water enhance the solubility of substrates or products. Although traditional water-miscible organic solvents also enhance solubility, they often inactivate enzymes, while ionic liquids do not. The enhanced solubility of substrates can increase the rate of reaction and often increases the regioor enantioselectivity. Ionic liquids can also be solvents for non-aqueous reactions. In these cases, they are especially suited to dissolve polar substrates. Polar organic solvent alternatives inactivate enzymes, but ionic liquids do not even when they have similar polarities. Besides their solubility properties, ionic liquids and deep eutectic solvents may be greener than organic solvents because ionic liquids are nonvolatile, and can be made from nontoxic components. This review covers selected examples of enzyme catalyzed reaction in ionic liquids that demonstrate their advantages and unique properties, and point out opportunities for new applications. Most examples involve hydrolases, but oxidoreductases and even whole cell reactions have been reported in ionic liquids.
Co-reporter:Qing Jing Dr. ;RomasJ. Kazlauskas Dr.
ChemCatChem 2010 Volume 2( Issue 8) pp:953-957
Publication Date(Web):
DOI:10.1002/cctc.201000159
Co-reporter:Qing Jing Dr. ;RomasJ. Kazlauskas Dr.
ChemCatChem 2010 Volume 2( Issue 8) pp:
Publication Date(Web):
DOI:10.1002/cctc.201090032
Co-reporter:Qing Jing Dr.;Krzysztof Okrasa Dr. ;RomasJ. Kazlauskas
Chemistry - A European Journal 2009 Volume 15( Issue 6) pp:1370-1376
Publication Date(Web):
DOI:10.1002/chem.200801673
Abstract
One useful synthetic reaction missing from nature's toolbox is the direct hydrogenation of substrates using hydrogen. Instead nature uses cofactors like NADH to reduce organic substrates, which adds complexity and cost to these reductions. To create an enzyme that can directly reduce organic substrates with hydrogen, researchers have combined metal hydrogenation catalysts with proteins. One approach is an indirect link where a ligand is linked to a protein and the metal binds to the ligand. Another approach is direct linking of the metal to protein, but nonspecific binding of the metal limits this approach. Herein, we report a direct hydrogenation of olefins catalyzed by rhodium(I) bound to carbonic anhydrase (CA-[Rh]). We minimized nonspecific binding of rhodium by replacing histidine residues on the protein surface using site-directed mutagenesis or by chemically modifying the histidine residues. Hydrogenation catalyzed by CA-[Rh] is slightly slower than for uncomplexed rhodium(I), but the protein environment induces stereoselectivity favoring cis- over trans-stilbene by about 20:1. This enzyme is the first cofactor-independent reductase that reduces organic molecules using hydrogen. This catalyst is a good starting point to create variants with tailored reactivity and selectivity. This strategy to insert transition metals in the active site of metalloenzymes opens opportunities to a wider range of enzyme-catalyzed reactions.
Co-reporter:Helge Jochens Dipl.-Biochem.;Konstanze Stiba Dr.;Christopher Savile Dr.;Ryota Fujii Dr.;Juin-Guo Yu;Tatsiana Gerassenkov;RomasJ. Kazlauskas ;UweT. Bornscheuer Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 19) pp:3532-3535
Publication Date(Web):
DOI:10.1002/anie.200806276
Co-reporter:Helge Jochens Dipl.-Biochem.;Konstanze Stiba Dr.;Christopher Savile Dr.;Ryota Fujii Dr.;Juin-Guo Yu;Tatsiana Gerassenkov;RomasJ. Kazlauskas Dr.;UweT. Bornscheuer Dr.
Angewandte Chemie 2009 Volume 121( Issue 19) pp:3584-3587
Publication Date(Web):
DOI:10.1002/ange.200806276
Co-reporter:Jae-Hoon Park;Hyun-Joon Ha ;Won Koo Lee ;Tobie Généreux-Vincent
ChemBioChem 2009 Volume 10( Issue 13) pp:2213-2222
Publication Date(Web):
DOI:10.1002/cbic.200900343
Abstract
Candida antarctica lipase B catalyzed the stereoselective ammoniolysis of N-alkyl aziridine-2-carboxylates in tBuOH saturated with ammonia and yielded the (2S)-aziridine-2-carboxamide and unreacted (2R)-aziridine-2-carboxylate. Varying the N-1 substituent on the aziridine ring changed the rate and stereoselectivity of the reaction. Substrates with a benzyl substituent or a (1′R)-1-phenylethyl substituent reacted approximately ten times faster than substrates with a (1′S)-1-phenylethyl substituent. Substrates with a benzyl substituent showed little stereoselectivity (E=5–7) while substrates with either a (1′R)- or (1′S)-1-phenylethyl substituent showed high stereoselectivity (D>50). Molecular modeling by using the current paradigm for enantioselectivity—binding of the slow enantiomer by an exchange-of-substituents orientation—could not account for the experimental results. However, modeling an umbrella-like-inversion orientation for the slow enantiomer could account for the experimental results. Steric hindrance between the methyl in the (1′S)-1-phenylethyl substituent and Thr138 and Ile189 in the acyl-binding site likely accounts for the slow reaction. Enantioselectivity likely stems from an unfavorable interaction of the methine hydrogen with Thr40 for the slow enantiomer and from subtle differences in the orientations of the other three substituents. This success in rationalizing the enantioselectivity supports the notion that an umbrella-like-inversion orientation can contribute to enantioselectivity in lipases.
Co-reporter:Jae-Hoon Park;Hyun-Joon Ha ;Won Koo Lee ;Tobie Généreux-Vincent
ChemBioChem 2009 Volume 10( Issue 13) pp:
Publication Date(Web):
DOI:10.1002/cbic.200990054
Co-reporter:Johnathan T. Gorke, Friedrich Srienc and Romas J. Kazlauskas
Chemical Communications 2008 (Issue 10) pp:1235-1237
Publication Date(Web):31 Jan 2008
DOI:10.1039/B716317G
Hydrolases show good catalytic activity in deep eutectic solvents, despite the presence of urea, which can denature enzymes, or alcohols, which can interfere with hydrolase-catalyzed reactions.
Co-reporter:PaulF. Mugford Dr.;Ulrike G. Wagner Dr.;Yun Jiang;Kurt Faber Dr.;RomasJ. Kazlauskas
Angewandte Chemie International Edition 2008 Volume 47( Issue 46) pp:8782-8793
Publication Date(Web):
DOI:10.1002/anie.200705159
Abstract
One often-cited weakness of biocatalysis is the lack of mirror-image enzymes for the formation of either enantiomer of a product in asymmetric synthesis. Enantiocomplementary enzymes exist as the solution to this problem in nature. These enzyme pairs, which catalyze the same reaction but favor opposite enantiomers, are not mirror-image molecules; however, they contain active sites that are functionally mirror images of one another. To create mirror-image active sites, nature can change the location of the binding site and/or the location of key catalytic groups. In this Minireview, X-ray crystal structures of enantiocomplementary enzymes are surveyed and classified into four groups according to how the mirror-image active sites are formed.
Co-reporter:Qing Jing
Chirality 2008 Volume 20( Issue 5) pp:724-735
Publication Date(Web):
DOI:10.1002/chir.20543
Abstract
Lipases show high enantioselectivity toward a wide range of secondary alcohols. An empirical rule based on the relative sizes of the substituents predicts which enantiomer reacts faster. X-ray structures of lipases provide a molecular basis for this empirical rule: their alcohol-binding pocket contains large hydrophobic pocket open to solvent and another smaller pocket. This predictable enantiopreference of lipases allows the determination of the absolute configuration of secondary alcohols using lipase-catalyzed kinetic resolution. Researchers have used this relative method to determine the configuration of ∼50 secondary alcohols either as the only method or in combination with other methods. Chirality, 2008. © 2008 Wiley-Liss, Inc.
Co-reporter:Christopher K. Savile;Romas J. Kazlauskas
Advanced Synthesis & Catalysis 2006 Volume 348(Issue 10-11) pp:
Publication Date(Web):19 JUL 2006
DOI:10.1002/adsc.200606040
Compared to an acetyl acyl group, the 3-(3-pyridine)propionyl group increases substrate binding to many proteases and substrate solubility in water, thereby increasing the rates of protease-catalyzed reactions. For example, proteases reacted up to six hundred-fold faster with the 3-(3-pyridine)propionyl ester of 1-phenylethanol than with the corresponding acetate ester. In addition, the 3-(3-pyridine)propionyl group enables a simple, mild acid extraction to separate the remaining starting material and product. To demonstrate the synthetic usefulness of this strategy, we resolved multi-gram quantities of (R)- and (S)-p-toluenesulfinamide with α-chymotrypsin and gram quantities of (R)- and (S)-2,2-dimethylcyclopentanol with subtilisin Carlsberg. The 3-(3-pyridyl)propionyl group was better for these resolutions than the corresponding acetate or dihydrocinnamate because it decreased the reaction time due to increased reactivity, decreased the reaction volume due to increased substrate solubility and enabled purification without chromatography. Molecular modeling suggests the enantioselectivity of α-chymotrypsin toward (R)-p-toluenesulfinamide is high (E=52) because of a favorable hydrophobic interaction between the p-tolyl group of the fast-reacting (R)-enantiomer and leaving group pocket. The enantioselectivity of subtilisin Carlsberg toward (S)-2,2-dimethylcyclopentanol is high (E=43) because the large substituent (the 2,2-dimethyl quaternary carbon) of the slow-reacting (R)-enantiomer cannot fit in the S1′ leaving group pocket.
Co-reporter:Iván Lavera Dr.;Susana Fernández Dr.;Julia Magdalena Dr.;Miguel Ferrero ;Harjap Grewal Dr.;Christopher K. Savile Dr. ;Vicente Gotor
ChemBioChem 2006 Volume 7(Issue 4) pp:
Publication Date(Web):21 FEB 2006
DOI:10.1002/cbic.200500451
Lipase from Pseudomonas cepacia (PCL) surprisingly favors acylation of the secondary hydroxyl at the 3′-position over the primary hydroxyl at the 5′-position in 2′-deoxynucleosides by up to >98:1. Catalytically productive tetrahedral intermediate analogues for both orientations were found by molecular modeling. However, acylation of the 3′-hydroxyl places the thymine base in the alternate hydrophobic pocket of PCL's substrate-binding site where it can hydrogen bond to the side-chain hydroxyls of Tyr23 and Tyr29 and the main chain carbonyl of Leu17. Conversely, acylation of the 5′-hydroxyl leaves the thymine base in the solvent where there is no favorable binding to the enzyme. We propose that these remote stabilizing interactions between the thymine base and PCL's substrate-binding site stabilize the 3′-acylation transition state and thus account for the unusual regioselectivity.
Co-reporter:Krzysztof Okrasa Dr.
Chemistry - A European Journal 2006 Volume 12(Issue 6) pp:
Publication Date(Web):17 JAN 2006
DOI:10.1002/chem.200501413
Carbonic anhydrase is a zinc metalloenzyme that catalyzes the hydration of carbon dioxide to bicarbonate. Replacing the active-site zinc with manganese yielded manganese-substituted carbonic anhydrase (CA[Mn]), which shows peroxidase activity with a bicarbonate-dependent mechanism. In the presence of bicarbonate and hydrogen peroxide, (CA[Mn]) catalyzed the efficient oxidation of o-dianisidine with kcat/KM=1.4×106 m−1 s−1, which is comparable to that for horseradish peroxidase, kcat/KM=57×106 m−1 s−1. CA[Mn] also catalyzed the moderately enantioselective epoxidation of olefins to epoxides (E=5 for p-chlorostyrene) in the presence of an amino-alcohol buffer, such as N,N-bis(2-hydroxyethyl)-2-aminoethanesulfonic acid (BES). This enantioselectivity is similar to that for natural heme-based peroxidases, but has the advantage that CA[Mn] avoids the formation of aldehyde side products. CA[Mn] degrades during the epoxidation limiting the yield of the epoxidations to <12 %. Replacement of active-site residues Asn62, His64, Asn67, Gln92, or Thr200 with alanine by site-directed mutagenesis decreased the enantioselectivity demonstrating that the active site controls the enantioselectivity of the epoxidation.
Co-reporter:Jeremy D. Cheeseman Dr.;Andrew D. Corbett Dr.;James L. Gleason
Chemistry - A European Journal 2005 Volume 11(Issue 6) pp:
Publication Date(Web):25 NOV 2004
DOI:10.1002/chem.200400371
Current drug discovery using combinatorial chemistry involves synthesis followed by screening, but emerging methods involve receptor-assistance to combine these steps. Adding stoichiometric amounts of receptor during library synthesis alters the kinetics or thermodynamics of the synthesis in a way that identifies the best-binding library members. Three main methods have emerged thus far in receptor-assisted combinatorial chemistry: dynamic combinatorial libraries, receptor-accelerated synthesis, and a new method, pseudo-dynamic libraries. Pseudo-dynamic libraries apply both thermodynamics and kinetics to amplify library members to easily observable levels, and attain selectivity heretofore unseen in receptor-assisted systems.
Co-reporter:Peter Bernhardt M.Sc.;Karl Hult Dr. Dr.
Angewandte Chemie International Edition 2005 Volume 44(Issue 18) pp:
Publication Date(Web):31 MAR 2005
DOI:10.1002/anie.200463006
Changing substrates: A mutation that forms a cis-proline–peptide bond in a loop structure close to the active site of an aryl esterase from Pseudomonas fluorescens converts the enzyme into a perhydrolase (see picture). The switch in activity is explained by a new hydrogen bond formed between a backbone carbonyl oxygen atom and the peroxy deacylation intermediate.
Co-reporter:Peter Bernhardt M.Sc.;Karl Hult Dr. Dr.
Angewandte Chemie 2005 Volume 117(Issue 18) pp:
Publication Date(Web):31 MAR 2005
DOI:10.1002/ange.200463006
Substratwechsel: Durch eine Mutation, die zu einer cis-Prolin-Peptidbindung in einer Schleifenstruktur nahe am aktiven Zentrum einer Arylesterase aus Pseudomonas fluorescens führt, wird aus dem Enzym eine Perhydrolase (siehe Bild). Das Umschalten der Aktivität wird mit einer neuen Wasserstoffbrücke zwischen einem Rückgrat-Carbonylsauerstoffatom und dem peroxidischen Desacylierungsintermediat erklärt.
Co-reporter:Iván Lavera Dr.;Susana Fernández Dr.;Julia Magdalena Dr.;Miguel Ferrero ;Vicente Gotor
ChemBioChem 2005 Volume 6(Issue 8) pp:
Publication Date(Web):24 JUN 2005
DOI:10.1002/cbic.200400422
Candida antarctica lipase B (CAL-B) catalyzes the regioselective acylation of natural thymidine with oxime esters and also the regioselective acylation of an analogue, 3′,5′-diamino-3′,5′-dideoxythymidine with nonactivated esters. In both cases, acylation favors the less hindered 5′-position over the 3′-position by upto 80-fold. Computer modeling of phosphonate transition-state analogues for the acylation of thymidine suggests that CAL-B favors acylation of the 5′-position because this orientation allows the thymine ring to bind in a hydrophobic pocket and forms stronger key hydrogen bonds than acylation of the 3′-position. On the other hand, computer modeling of phosphonamidate analogues of the transition states for acylation of either the 3′- or 5′-amino groups in 3′,5′-diamino-3′,5′-dideoxythymidine shows similar orientations and hydrogen bonds and, thus, does not explain the high regioselectivity. However, computer modeling of inverse structures, in which the acyl chain binds in the nucleophile pocket and vice versa, does rationalize the observed regioselectivity. The inverse structures fit the 5′-, but not the 3′-intermediate thymine ring, into the hydrophobic pocket, and form a weak new hydrogen bond between the O-2 carbonyl atom of the thymine and the nucleophile amine only for the 5′-intermediate. A water molecule might transfer a proton from the ammonium group to the active-site histidine. As a test of this inverse orientation, we compared the acylation of thymidine and 3′,5′-diamino-3′,5′-dideoxythymidine with butyryl acyl donors and with isosteric methoxyacetyl acyl donors. Both acyl donors reacted at equal rates with thymidine, but the methoxyacetyl acyl donor reacted four times faster than the butyryl acyl donor with 3′,5′-diamino-3′,5′-dideoxythymidine. This faster rate is consistent with an inverse orientation for 3′,5′-diamino-3′,5′-dideoxythymidine, in which the ether oxygen atom of the methoxyacetyl group can form a similar hydrogen bond to the nucleophilic amine. This combination of modeling and experiments suggests that such lipase-catalyzed reactions of apparently close substrate analogues like alcohols and amines might follow different pathways.
Co-reporter:Uwe T. Bornscheuer Dr. Dr.
Angewandte Chemie 2004 Volume 116(Issue 45) pp:
Publication Date(Web):2 NOV 2004
DOI:10.1002/ange.200460416
Mit der Entdeckung hochstereoselektiver Enzyme mit breiter Substratspezifität hat sich die Biokatalyse in den letzten Jahrzehnten rasant entwickelt. Ein neues Forschungsgebiet beschäftigt sich mit Enzymen, die eine breite Reaktionsspezifität bei der Katalyse alternativer Reaktionen aufweisen. Oft unterschätzt, spielt diese katalytische Promiskuität (catalytic promiscuity) eine natürliche Rolle in der Evolution und in manchen Fällen auch in der Biosynthese von Sekundärmetaboliten. In diesem Kurzaufsatz werden Beispiele für katalytische Promiskuität mit aktuellen und möglichen Anwendungen in der Synthese vorgestellt. Kombiniert mit Protein-Engineering könnten dank dieser Eigenschaft die Verwendungsmöglichkeiten von Enzymen in der organischen Synthese deutlich erweitert werden.
Co-reporter:Andrew D. Corbett;Jeremy D. Cheeseman ;James L. Gleason
Angewandte Chemie International Edition 2004 Volume 43(Issue 18) pp:
Publication Date(Web):31 MAR 2004
DOI:10.1002/anie.200453769
Enzyme protection: An irreversible solid-phase, aqueous peptide coupling resulted in the formation of a library of eight dipeptides, while an irreversible protease-catalyzed hydrolysis destroyed them. Those dipeptides that bound to carbonic anhydrase were protected from destructions. Six cycles of active ester addition produced only the best-binding dipeptide (>100:1) in 29 % yield.
Co-reporter:Andrew D. Corbett;Jeremy D. Cheeseman ;James L. Gleason
Angewandte Chemie 2004 Volume 116(Issue 18) pp:
Publication Date(Web):31 MAR 2004
DOI:10.1002/ange.200453769
Schutz vor Enzymen: Eine irreversible wässrige Festphasenpeptidkupplung führte zu einer Bibliothek aus acht Dipeptiden, die durch irreversible proteasekatalysierte Hydrolyse wieder zerstört werden. Dipeptide, die einen Komplex mit Carbonsäureanhydrase bilden, sind gegen Abbau geschützt. Durch wiederholte Addition des aktivierten Esters wurde nach sechs Zyklen ausschließlich das am stärksten koordinierende Dipeptid (>100:1) in 29 % Ausbeute erhalten.
Co-reporter:Paul F. Mugford;Susan M. Lait;Brian A. Keay Dr. Dr.
ChemBioChem 2004 Volume 5(Issue 7) pp:
Publication Date(Web):1 JUL 2004
DOI:10.1002/cbic.200300909
cis,cis-(±)-6-(2,2-Dimethylpropanamido)spiro[4.4]nonan-1-ol, 1, a chiral auxiliary for Diels–Alder additions, was resolved by enzyme-catalyzed hydrolysis of the corresponding butyrate and acrylate esters. Subtilisin Carlsberg protease and bovine cholesterol esterase both showed high enantioselectivity in this process, but favored opposite enantiomers. Subtilisin Carlsberg favored esters of (1S,5S,6S)-1, while bovine cholesterol esterase favored esters of (1R,5R,6R)-1, consistent with the approximately mirror-image arrangement of the active sites of subtilisins and lipases/esterases. A gram-scale resolution of 1-acrylate with subtilisin Carlsberg yielded (1S,5S,6S)-1 (1.1 g, 46 % yield, 99 % ee) and (1R,5R,6R)-1-acrylate (1.3 g, 44 % yield, 99 % ee) although the reaction was slow. The high enantioselectivity combined with the conformational rigidity of the substrate made this an ideal example to identify the molecular basis of the enantioselectivity of subtilisin Carlsberg toward secondary alcohols. When modeled, the favored (1S,5S,6S) enantiomer adopted a catalytically productive conformation with two longer-than-expected hydrogen bonds, consistent with the slow reaction rate. The unfavored (1R,5R,6R) enantiomer encountered severe steric interactions with catalytically essential residues in the model. It either distorted the catalytic histidine position or encountered severe steric strain with Asn155, an oxyanion-stabilizing residue.
Co-reporter:Neil A. Somers, Romas J. Kazlauskas
Tetrahedron: Asymmetry 2004 Volume 15(Issue 18) pp:2991-3004
Publication Date(Web):20 September 2004
DOI:10.1016/j.tetasy.2004.07.044
To identify potential applications of nineteen esterases from thermophiles, we mapped their substrate selectivity and enantioselectivity using a library of 50 esters. We measured the selectivities colorimetrically using Quick E, which uses pH indicators to detect hydrolysis and a chromogenic reference compound as an internal control. The substrate selectivity mapping revealed one esterase, E018b, with a strong preference for acetyl esters (14- to 25-fold over hexanoate). The enantioselectivity mapping revealed a number of cases of high enantioselectivity. Thirteen of the 19 esterases showed moderate or better enantioselectivity (>19) toward 1-phenethyl butyrate favoring the (R)-enantiomer and two esterases (E008, E013) showed moderate or better enantioselectivity (>20) toward methyl 2-chloropropionate favoring the (S)-enantiomer. Three esterases (E001, E004, E005) showed high (>46) enantioselectivity toward menthyl acetate favoring the (R)-enantiomer. This rapid mapping of the selectivity simplifies the characterization of new enzymes.
Co-reporter:Uwe T. Bornscheuer Dr. Dr.
Angewandte Chemie International Edition 2004 Volume 43(Issue 45) pp:
Publication Date(Web):2 NOV 2004
DOI:10.1002/anie.200460416
Biocatalysis has expanded rapidly in the last decades with the discoveries of highly stereoselective enzymes with broad substrate specificity. A new frontier for biocatalysis is broad reaction specificity, where enzymes catalyze alternate reactions. Although often underappreciated, catalytic promiscuity has a natural role in evolution and occasionally in the biosynthesis of secondary metabolites. Examples of catalytic promiscuity with current or potential applications in synthesis are reviewed here. Combined with protein engineering, the catalytic promiscuity of enzymes may broadly extend their usefulness in organic synthesis.
Co-reporter:Alessandra Mezzetti, Curtis Keith, Romas J. Kazlauskas
Tetrahedron: Asymmetry 2003 Volume 14(Issue 24) pp:3917-3924
Publication Date(Web):12 December 2003
DOI:10.1016/j.tetasy.2003.09.049
Although lipase from Pseudomonas cepacia (PCL) shows high enantioselectivity towards many secondary alcohols, it usually exhibits only low to moderate enantioselectivity towards primary alcohols. To increase this enantioselectivity, we optimised the reaction conditions for the PCL-catalysed hydrolysis of esters of three chiral primary alcohols: 2-methyl-3-phenyl-1-propanol 1, 2-phenoxy-1-propanol 2 and solketal 3. The enantioselectivity towards 1-acetate increased from E=16 to 38 upon changing the solvent from ethyl ether/phosphate buffer to 30% n-propanol in phosphate buffer and increased again to E ≥190 upon changing the substrate from 1-acetate to 1-heptanoate. The same changes increased the enantioselectivity towards alcohol 2 from E=17 to 70, but did not significantly increase the enantioselectivity towards alcohol 3. The best solvent was similar to the solvent used to crystallise the open form of PCL and likely stabilises the open form of PCL. This stabilisation may increase the enantioselectivity by removing kinetic contributions from a non-enantioselective lid-opening step. We determined the kinetic contribution of the lid-opening step by measuring the interfacial activation of PCL. The activation energy for the PCL-catalysed hydrolysis of ethyl acetate was at least 2.6 kcal/mol lower in the presence of a water–organic solvent interface.Graphic(S)-(−)-2-Methyl-3-phenyl-1-propanolC10H14OEe=98%[α]D20=−11.3 (c 0.124, C6H6)Source of chirality: enzymatic kinetic resolutionAbsolute configuration: S
Co-reporter:Guo-Qiang Chen, Romas Kazlauskas
Current Opinion in Biotechnology (December 2011) Volume 22(Issue 6) pp:747-748
Publication Date(Web):1 December 2011
DOI:10.1016/j.copbio.2011.08.003
Co-reporter:Johnathan T. Gorke, Friedrich Srienc and Romas J. Kazlauskas
Chemical Communications 2008(Issue 10) pp:NaN1237-1237
Publication Date(Web):2008/01/31
DOI:10.1039/B716317G
Hydrolases show good catalytic activity in deep eutectic solvents, despite the presence of urea, which can denature enzymes, or alcohols, which can interfere with hydrolase-catalyzed reactions.





![2,4-Pentanedione, 3-[(1R)-2-nitro-1-phenylethyl]-](http://img.cochemist.com/ccimg/190000/189952-21-4.png)
![2,4-Pentanedione, 3-[(1R)-2-nitro-1-phenylethyl]-](http://img.cochemist.com/ccimg/190000/189952-21-4_b.png)
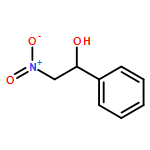
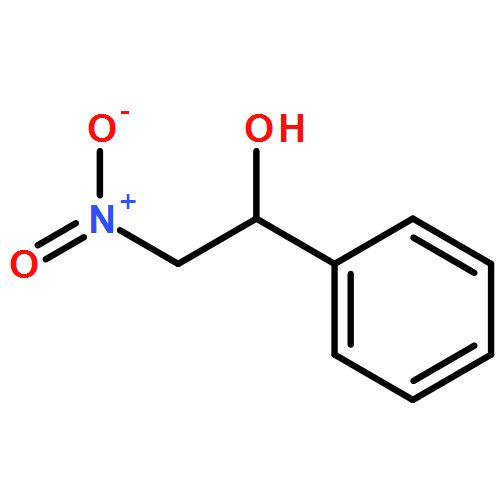
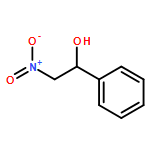
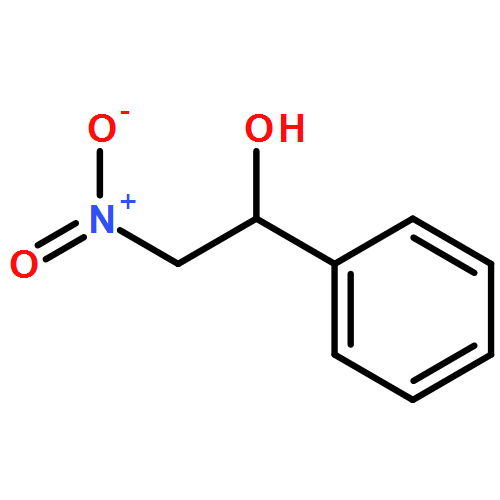
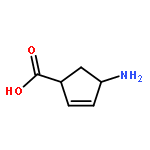
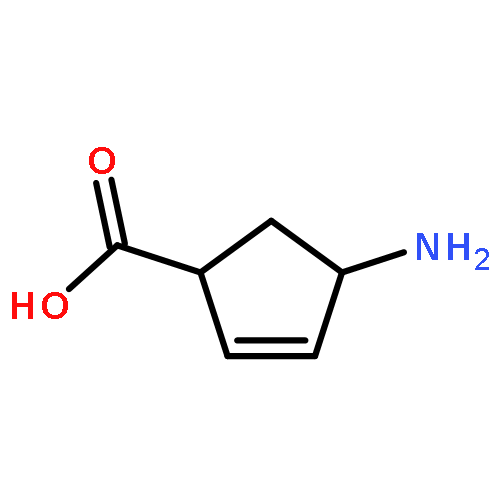


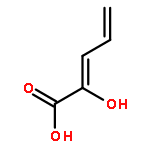

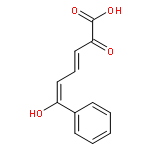



![L-Cysteine,S-[(2R)-2-amino-2-carboxyethyl]-](http://img.cochemist.com/ccimg/1000/922-55-4.png)
![L-Cysteine,S-[(2R)-2-amino-2-carboxyethyl]-](http://img.cochemist.com/ccimg/1000/922-55-4_b.png)



