Co-reporter:Lewis D. Turner, Abbey J. Summers, Laura O. Johnson, Margaret A. Knowles, and Colin W. G. Fishwick
ACS Medicinal Chemistry Letters December 14, 2017 Volume 8(Issue 12) pp:1264-1264
Publication Date(Web):November 11, 2017
DOI:10.1021/acsmedchemlett.7b00349
Structure-based drug design (SBDD) has become a powerful tool utilized by medicinal chemists to rationally guide the drug discovery process. Herein, we describe the use of SPROUT, a de novo-based program, to identify an indazole-based pharmacophore for the inhibition of fibroblast growth factor receptor (FGFR) kinases, which are validated targets for cancer therapy. Hit identification using SPROUT yielded 6-phenylindole as a small fragment predicted to bind to FGFR1. With the aid of docking models, several modifications to the indole were made to optimize the fragment to an indazole-containing pharmacophore, leading to a library of compounds containing 23 derivatives. Biological evaluation revealed that these indazole-containing fragments inhibited FGFR1–3 in the range of 0.8–90 μM with excellent ligand efficiencies of 0.30–0.48. Some compounds exhibited moderate selectivity toward individual FGFRs, indicating that further optimization using SBDD may lead to potent and selective inhibitors of the FGFR family.Keywords: de novo; FGFR; fragment; indazole; SBDD;
Co-reporter:Ronald Grigg, Elghareeb E. Elboray, Sunisa Akkarasamiyo, Nutthawat Chuanopparat, H. Ali Dondas, Hussien H. Abbas-Temirek, Colin W. G. Fishwick, Moustafa F. Aly, Boonsong Kongkathip and Ngampong Kongkathip
Chemical Communications 2016 vol. 52(Issue 1) pp:164-166
Publication Date(Web):21 Oct 2015
DOI:10.1039/C5CC07526B
Palladium catalysed three component cascade process, involving coupling of 2-iodobenzoates, -benzaldehydes, or acetophenones with substituted allenes and ammonium tartrate as an ammonium surrogate, provides a novel and facile route to substituted functionalised isoquinolinones and isoquinolines in good yields.
Co-reporter:H. Ali Dondas, Aiden Hempshall, Sarah Narramore, Colin Kilner, Colin W.G. Fishwick, Ronald Grigg
Tetrahedron 2016 Volume 72(Issue 10) pp:1316-1329
Publication Date(Web):10 March 2016
DOI:10.1016/j.tet.2016.01.025
γ-Carbolines were prepared from pyrido[4,3-b]-5H-indoles via Pd(0)-catalysed, stereo and regioselective allene/uridine allene insertion 3- and 5-component cascades. This versatile reaction sequence gives a range of structurally diverse carboline derivatives and tolerates a broad range of substrates. The power of this approach has been harnessed to produce γ-carboline based HDAC inhibitors.
Co-reporter:Craig A. Avery, Richard J. Pease, Kerrie Smith, May Boothby, Helen M. Buckley, Peter J. Grant, Colin W.G. Fishwick
European Journal of Medicinal Chemistry 2015 Volume 98() pp:49-53
Publication Date(Web):15 June 2015
DOI:10.1016/j.ejmech.2015.05.019
•Inhibitors of human blood clotting FXIII-A were designed using molecular modelling methods.•Synthesis of a range of these cis-bisamido epoxides is described.•Inhibitors with IC50s as low as 4 nM against FXIII-A were evaluated.•Inhibitors were stable in the presence of thiols.A novel class of potent FXIII-A inhibitors containing a (±) cis-bisamido epoxide pharmacophore is described. The compounds display highly potent inhibition of FXIII-A (IC50 = 5–500 nM) in an in vitro assay. In contrast to other types of previously described covalent transglutaminase inhibitors, the bis-amido epoxides exhibited no measurable reactivity with glutathione, therefore possibly rendering this class of compounds suitable for future in vivo investigations. Additionally, the compounds show selective inhibition for FXIII-A against the cysteine protease, cathepsin S although they proved to have similar potency with a closely related transglutaminase, TGII, to that observed for FXIII-A.
Co-reporter:Ian A. Yule, Lloyd G. Czaplewski, Stephanie Pommier, David T. Davies, Sarah K. Narramore, Colin W.G. Fishwick
European Journal of Medicinal Chemistry 2014 Volume 86() pp:31-38
Publication Date(Web):30 October 2014
DOI:10.1016/j.ejmech.2014.08.025
•Inhibitors of bacterial DNA Gyrase were designed using de novo computational methods.•Synthesis of a range of these pyridine-3-carboxamide-6-yl-ureas is described.•Inhibitors with IC50s as low as 24 nM against DNA Gyrase were evaluated.•Inhibitors were active against a range of Gram positive bacteria including Staphylococcus aureus.The development of antibacterial drugs based on novel chemotypes is essential to the future management of serious drug resistant infections. We herein report the design, synthesis and SAR of a novel series of N-ethylurea inhibitors based on a pyridine-3-carboxamide scaffold targeting the ATPase sub-unit of DNA gyrase. Consideration of structural aspects of the GyrB ATPase site has aided the development of this series resulting in derivatives that demonstrate excellent enzyme inhibitory activity coupled to potent Gram positive antibacterial efficacy.
Co-reporter:Ricky Cain, Sarah Narramore, Martin McPhillie, Katie Simmons, Colin W.G. Fishwick
Bioorganic Chemistry 2014 Volume 55() pp:69-76
Publication Date(Web):August 2014
DOI:10.1016/j.bioorg.2014.05.008
•Structure-based drug design is an important tool in the design of antibacterial compounds.•Use of computational design in the modification of known inhibitors.•Use of SPROUT, CAVEAT and LUDI for the de novo design of inhibitors of bacterial targets.•vHTS using AutoDock, eHiTs, Glide and FRED to identify potential antibacterial compounds.In recent years bacterial resistance has been observed against many of our current antibiotics, for instance most worryingly against the cephalosporins which are typically the last line of defence against many bacterial infections. Additionally the failure of high throughput screening in the discovery of new antibacterial drug leads has led to a decline in the number of antibacterial agents reaching the market. Alternative methods of drug discovery including structure based drug design are needed to meet the threats caused by the emergence of resistance. In this review we explore the latest advancements in the identification of new antibacterial agents through the use of a number of structure based drug design programs.
Co-reporter:N. Yi Mok ; James Chadwick ; Katherine A. B. Kellett ; Eva Casas-Arce ; Nigel M. Hooper ; A. Peter Johnson
Journal of Medicinal Chemistry 2013 Volume 56(Issue 5) pp:1843-1852
Publication Date(Web):February 1, 2013
DOI:10.1021/jm301127x
β-Secretase (BACE1), the enzyme responsible for the first and rate-limiting step in the production of amyloid-β peptides, is an attractive target for the treatment of Alzheimer’s disease. In this study, we report the application of the de novo fragment-based molecular design program SPROUT to the discovery of a series of nonpeptide BACE1 inhibitors based upon a biphenylacetamide scaffold. The binding affinity of molecules based upon this designed molecular scaffold was increased from an initial BACE1 IC50 of 323 μM to 27 μM following the synthesis of a library of optimized ligands whose structures were refined using the recently developed SPROUT-HitOpt software. Although a number of inhibitors were found to exhibit cellular toxicity, one compound in the series was found to have useful BACE1 inhibitory activity in a cellular assay with minimal cellular toxicity. This work demonstrates the power of an in silico fragment-based molecular design approach in the discovery of novel BACE1 inhibitors.
Co-reporter:Paul T. P. Bedingfield ; Deborah Cowen ; Paul Acklam ; Fraser Cunningham ; Mark R. Parsons ; Glenn A. McConkey ; Colin W. G. Fishwick ;A. Peter Johnson
Journal of Medicinal Chemistry 2012 Volume 55(Issue 12) pp:5841-5850
Publication Date(Web):May 23, 2012
DOI:10.1021/jm300157n
The de novo pyrimidine biosynthesis enzyme dihydroorotate dehydrogenase is an emerging drug target for the treatment of malaria. In this context a key property of Plasmodium falciparum DHODH (PfDHODH) is that it can be selectively inhibited over its human homologue (HsDHODH). However, HsDHODH is also a validated drug target for autoimmune diseases such as arthritis. Here a series of novel inhibitors is described that includes compounds that switch specificity between the two enzymes as a result of small alterations in chemical structure. Structure–activity relationship (SAR), crystallography, docking, and mutagenesis studies are used to examine the binding modes of the compounds within the two enzymes and to reveal structural changes induced by inhibitor binding. Within this series, compounds with therapeutically relevant HsDHODH activity are described and their binding modes characterized using X-ray crystallography, which reveals a novel conformational shift within the inhibitor binding site.
Co-reporter:Martin J. McPhillie, Rachel Trowbridge, Katherine R. Mariner, Alex J. O’Neill, A. Peter Johnson, Ian Chopra, and Colin W. G. Fishwick
ACS Medicinal Chemistry Letters 2011 Volume 2(Issue 10) pp:729
Publication Date(Web):July 29, 2011
DOI:10.1021/ml200087m
Bacterial RNA polymerase (RNAP) is essential for transcription and is an antibacterial target for small molecule inhibitors. The binding region of myxopyronin B (MyxB), a bacterial RNAP inhibitor, offers the possibility of new inhibitor design. The molecular design program SPROUT has been used in conjunction with the X-ray cocrystal structure of Thermus thermophilus RNAP with MyxB to design novel inhibitors based on a substituted pyridyl-benzamide scaffold. A series of molecules, with molecular masses <350 Da, have been prepared using a simple synthetic approach. A number of these compounds inhibited Escherichia coli RNAP.Keywords: Bacterial RNA polymerase; myxopyronin B; small molecule inhibitors; structure-based ligand discovery
Co-reporter:Matthew Davies ; Timo Heikkilä ; Glenn A. McConkey ; Colin W. G. Fishwick ; Mark R. Parsons ;A. Peter Johnson
Journal of Medicinal Chemistry 2009 Volume 52(Issue 9) pp:2683-2693
Publication Date(Web):April 7, 2009
DOI:10.1021/jm800963t
Pyrimidine biosynthesis is an attractive drug target in a variety of organisms, including humans and the malaria parasite Plasmodium falciparum. Dihydroorotate dehydrogenase, an enzyme catalyzing the only redox reaction of the pyrimidine biosynthesis pathway, is a well-characterized target for chemotherapeutical intervention. In this study, we have applied SPROUT-LeadOpt, a software package for structure-based drug discovery and lead optimization, to improve the binding of the active metabolite of the anti-inflammatory drug leflunomide to the target cavities of the P. falciparum and human dihydroorotate dehydrogenases. Following synthesis of a library of compounds based upon the SPROUT-optimized molecular scaffolds, a series of inhibitors generally showing good inhibitory activity was obtained, in keeping with the SPROUT-LeadOpt predictions. Furthermore, cocrystal structures of five of these SPROUT-designed inhibitors bound in the ubiquinone binding cavity of the human dihydroorotate dehydrogenase are also analyzed.
Co-reporter:N. Yi Mok, James Chadwick, Katherine A.B. Kellett, Nigel M. Hooper, A. Peter Johnson, Colin W.G. Fishwick
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 23) pp:6770-6774
Publication Date(Web):1 December 2009
DOI:10.1016/j.bmcl.2009.09.103
A novel series of isatin-based inhibitors of β-secretase (BACE-1) have been identified using a virtual high-throughput screening approach. Structure–activity relationship studies revealed structural features important for inhibition. Docking studies suggest these inhibitors may bind within the BACE-1 active site through H-bonding interactions involving the catalytic aspartate residues.A novel series of isatin-based inhibitors of β-secretase (BACE-1) have been identified using a virtual high-throughput screening approach. Structure–activity relationship studies revealed structural features important for inhibition. Docking studies suggest these inhibitors may bind within the BACE-1 active site through H-bonding interactions involving the catalytic aspartate residues.
Co-reporter:Gilbert E. Besong;Julieanne M. Bostock Dr.;Will Stubbings Dr.;Ian Chopra Dr.;David I. Roper Dr.;Adrian J. Lloyd Dr. Dr.;A. Peter Johnson Dr.
Angewandte Chemie 2005 Volume 117(Issue 39) pp:
Publication Date(Web):13 SEP 2005
DOI:10.1002/ange.200501662
Blaupausen für einen Inhibitor: Bei der Suche nach einem neuartigen Templat für enzymselektive Inhibitoren wurde das Moleküldesignprogramm SPROUT mit Röntgenstrukturdaten der bakteriellen Enzyme DdlB und VanA kombiniert. Eine kurze und effiziente Synthese lieferte einen Inhibitor, der – wie vorhergesagt – eine enzymselektive inhibitorische Wirkung zeigte.
Co-reporter:Gilbert E. Besong, Julieanne M. Bostock, Will Stubbings, Ian Chopra, David I. Roper, Adrian J. Lloyd, Colin W. G. Fishwick,A. Peter Johnson
Angewandte Chemie International Edition 2005 44(39) pp:
Publication Date(Web):
DOI:10.1002/anie.200501662
Co-reporter:Joseph Shaw, Mark Harris, Colin W.G. Fishwick
Antiviral Research (December 2015) Volume 124() pp:54-60
Publication Date(Web):December 2015
DOI:10.1016/j.antiviral.2015.10.001
Co-reporter:Ronald Grigg, Elghareeb E. Elboray, Sunisa Akkarasamiyo, Nutthawat Chuanopparat, H. Ali Dondas, Hussien H. Abbas-Temirek, Colin W. G. Fishwick, Moustafa F. Aly, Boonsong Kongkathip and Ngampong Kongkathip
Chemical Communications 2016 - vol. 52(Issue 1) pp:NaN166-166
Publication Date(Web):2015/10/21
DOI:10.1039/C5CC07526B
Palladium catalysed three component cascade process, involving coupling of 2-iodobenzoates, -benzaldehydes, or acetophenones with substituted allenes and ammonium tartrate as an ammonium surrogate, provides a novel and facile route to substituted functionalised isoquinolinones and isoquinolines in good yields.

![3H-Pyrrolo[1,2-a]indole-1,2-dicarboxylic acid, 9,9a-dihydro-9,9,9a-trimethyl-, dimethyl ester](http://img.cochemist.com/ccimg/114700/114676-05-0.png)
![3H-Pyrrolo[1,2-a]indole-1,2-dicarboxylic acid, 9,9a-dihydro-9,9,9a-trimethyl-, dimethyl ester](http://img.cochemist.com/ccimg/114700/114676-05-0_b.png)
![Acetic acid, [(2-phenyl-1-thioxoethyl)thio]-](http://img.cochemist.com/ccimg/53000/52999-90-3.png)
![Acetic acid, [(2-phenyl-1-thioxoethyl)thio]-](http://img.cochemist.com/ccimg/53000/52999-90-3_b.png)
![Silane, (1,1-dimethylethyl)dimethyl[(phenylthio)methoxy]-](http://img.cochemist.com/ccimg/94800/94718-51-1.png)
![Silane, (1,1-dimethylethyl)dimethyl[(phenylthio)methoxy]-](http://img.cochemist.com/ccimg/94800/94718-51-1_b.png)
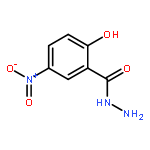
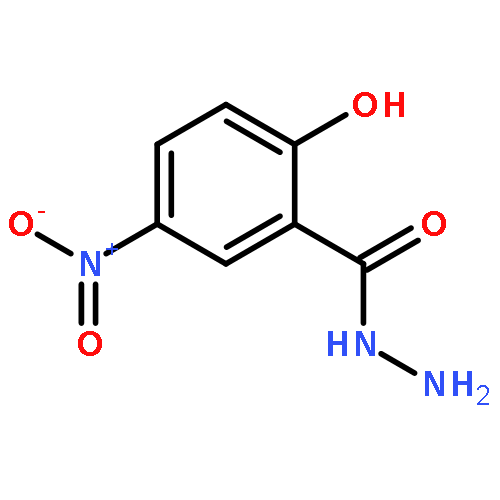
![Benzoic acid, 2-[([1,1'-biphenyl]-4-ylcarbonyl)amino]-](http://img.cochemist.com/ccimg/94600/94578-69-5.png)
![Benzoic acid, 2-[([1,1'-biphenyl]-4-ylcarbonyl)amino]-](http://img.cochemist.com/ccimg/94600/94578-69-5_b.png)
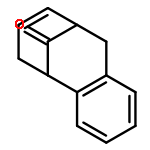
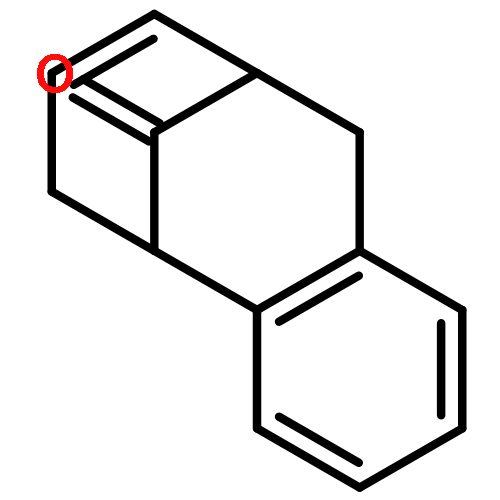
![[1,1'-Biphenyl]-4-acetic acid, 3-methoxy-](http://img.cochemist.com/ccimg/5100/5001-97-8.png)
![[1,1'-Biphenyl]-4-acetic acid, 3-methoxy-](http://img.cochemist.com/ccimg/5100/5001-97-8_b.png)

![BENZOIC ACID, 2-[METHYL(2-NAPHTHALENYLCARBONYL)AMINO]-](http://img.cochemist.com/ccimg/74500/74480-77-6.png)
![BENZOIC ACID, 2-[METHYL(2-NAPHTHALENYLCARBONYL)AMINO]-](http://img.cochemist.com/ccimg/74500/74480-77-6_b.png)

![Benzo[b]thiophene, 2,3-dihydro-3-[(4-methylphenyl)thio]-, 1,1-dioxide](http://img.cochemist.com/ccimg/246200/246169-01-7.png)
![Benzo[b]thiophene, 2,3-dihydro-3-[(4-methylphenyl)thio]-, 1,1-dioxide](http://img.cochemist.com/ccimg/246200/246169-01-7_b.png)

![Benzoic acid, 2-[(2-naphthalenylcarbonyl)amino]-](http://img.cochemist.com/ccimg/106700/106696-36-0.png)
![Benzoic acid, 2-[(2-naphthalenylcarbonyl)amino]-](http://img.cochemist.com/ccimg/106700/106696-36-0_b.png)

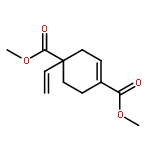
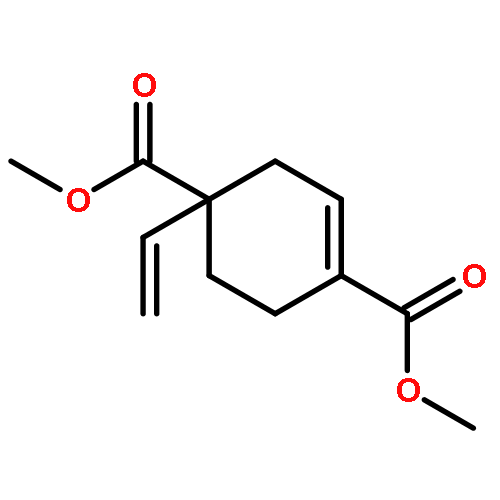


![Acetamide, N-[1,1'-biphenyl]-4-yl-2-cyano-](http://img.cochemist.com/ccimg/168200/168152-04-3.png)
![Acetamide, N-[1,1'-biphenyl]-4-yl-2-cyano-](http://img.cochemist.com/ccimg/168200/168152-04-3_b.png)




![3H-Diazirine, 3-[4-(methylsulfonyl)phenyl]-3-(trifluoromethyl)-](http://img.cochemist.com/ccimg/150800/150772-82-0.png)
![3H-Diazirine, 3-[4-(methylsulfonyl)phenyl]-3-(trifluoromethyl)-](http://img.cochemist.com/ccimg/150800/150772-82-0_b.png)
![POTASSIUM CYANATE, [14C]](http://img.cochemist.com/ccimg/67900/67877-97-8.png)
![POTASSIUM CYANATE, [14C]](http://img.cochemist.com/ccimg/67900/67877-97-8_b.png)
![methyl [(1E)-5-{6-hydroxy-5-[(2E,4E,9Z,13E)-8-hydroxy-2,5,9-trimethylpentadeca-2,4,9,13-tetraenoyl]-4-oxo-4H-pyran-2-yl}hex-1-en-1-yl]carbamate](http://img.cochemist.com/ccimg/93200/93195-32-5.png)
![methyl [(1E)-5-{6-hydroxy-5-[(2E,4E,9Z,13E)-8-hydroxy-2,5,9-trimethylpentadeca-2,4,9,13-tetraenoyl]-4-oxo-4H-pyran-2-yl}hex-1-en-1-yl]carbamate](http://img.cochemist.com/ccimg/93200/93195-32-5_b.png)

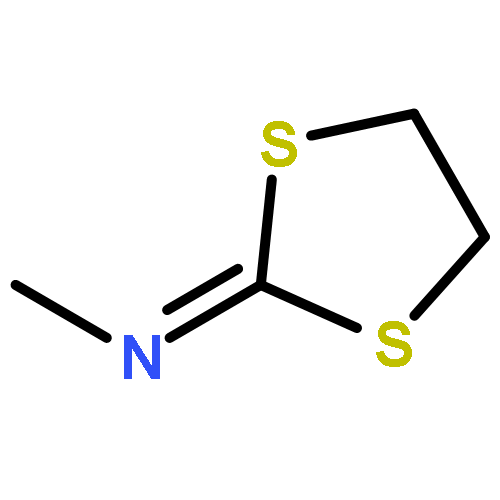
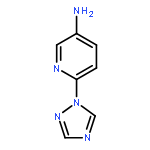



![2-(Benzylthio)-5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol](http://img.cochemist.com/ccimg/51700/51646-33-4.png)
![2-(Benzylthio)-5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol](http://img.cochemist.com/ccimg/51700/51646-33-4_b.png)
![1-(4-Chlorobenzyl)-1H-benzo[d]imidazole](http://img.cochemist.com/ccimg/107500/107456-42-8.png)
![1-(4-Chlorobenzyl)-1H-benzo[d]imidazole](http://img.cochemist.com/ccimg/107500/107456-42-8_b.png)





![{[2-amino-4-hydroxy-4-(4-hydroxyphenyl)-3-methylbutanoyl]amino}[5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3,4-dihydroxytetrahydrofuran-2-yl]acetic acid (non-preferred name)](http://img.cochemist.com/ccimg/74400/74342-20-4.png)
![{[2-amino-4-hydroxy-4-(4-hydroxyphenyl)-3-methylbutanoyl]amino}[5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3,4-dihydroxytetrahydrofuran-2-yl]acetic acid (non-preferred name)](http://img.cochemist.com/ccimg/74400/74342-20-4_b.png)
![[1,1'-Biphenyl]-4-amine, 3-chloro-](http://img.cochemist.com/ccimg/7300/7285-66-7.png)
![[1,1'-Biphenyl]-4-amine, 3-chloro-](http://img.cochemist.com/ccimg/7300/7285-66-7_b.png)
![Benzo[b]thiophene, 3-phenyl-](http://img.cochemist.com/ccimg/14400/14315-12-9.png)
![Benzo[b]thiophene, 3-phenyl-](http://img.cochemist.com/ccimg/14400/14315-12-9_b.png)
![[1,1'-Biphenyl]-4-amine, 2,2'-dichloro-](http://img.cochemist.com/ccimg/102900/102871-33-0.png)
![[1,1'-Biphenyl]-4-amine, 2,2'-dichloro-](http://img.cochemist.com/ccimg/102900/102871-33-0_b.png)
![1-Propanol, 3-[(triphenylmethyl)thio]-](http://img.cochemist.com/ccimg/100400/100360-56-3.png)
![1-Propanol, 3-[(triphenylmethyl)thio]-](http://img.cochemist.com/ccimg/100400/100360-56-3_b.png)
![9,9'-[(2R,3R,3aS,7aR,9R,10R,10aS,14aR)-3,5,10,12-tetrahydroxy-5,12-dioxidooctahydro-2H,7H-difuro[3,2-d:3',2'-j][1,3,7,9,2,8]tetraoxadiphosphacyclododecine-2,9-diyl]bis(2-amino-3,9-dihydro-6H-purin-6-one)](http://img.cochemist.com/ccimg/61100/61093-23-0.png)
![9,9'-[(2R,3R,3aS,7aR,9R,10R,10aS,14aR)-3,5,10,12-tetrahydroxy-5,12-dioxidooctahydro-2H,7H-difuro[3,2-d:3',2'-j][1,3,7,9,2,8]tetraoxadiphosphacyclododecine-2,9-diyl]bis(2-amino-3,9-dihydro-6H-purin-6-one)](http://img.cochemist.com/ccimg/61100/61093-23-0_b.png)







![3,6-Dioxabicyclo[3.1.0]hexane-2,4-dione(9CI)](http://img.cochemist.com/ccimg/16200/16191-17-6.png)
![3,6-Dioxabicyclo[3.1.0]hexane-2,4-dione(9CI)](http://img.cochemist.com/ccimg/16200/16191-17-6_b.png)








![L-Phenylalanine, 4-[3-(trifluoromethyl)-3H-diazirin-3-yl]-](http://img.cochemist.com/ccimg/92400/92367-16-3.png)
![L-Phenylalanine, 4-[3-(trifluoromethyl)-3H-diazirin-3-yl]-](http://img.cochemist.com/ccimg/92400/92367-16-3_b.png)




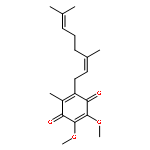


![(3S,3aR,4S,6S,6aR,7S,8S,9bS)-6-(acetyloxy)-4-(butanoyloxy)-3,3a-dihydroxy-3,6,9-trimethyl-8-{[(2Z)-2-methylbut-2-enoyl]oxy}-2-oxo-2,3,3a,4,5,6,6a,7,8,9b-decahydroazuleno[4,5-b]furan-7-yl octanoate](http://img.cochemist.com/ccimg/67600/67526-95-8.png)
![(3S,3aR,4S,6S,6aR,7S,8S,9bS)-6-(acetyloxy)-4-(butanoyloxy)-3,3a-dihydroxy-3,6,9-trimethyl-8-{[(2Z)-2-methylbut-2-enoyl]oxy}-2-oxo-2,3,3a,4,5,6,6a,7,8,9b-decahydroazuleno[4,5-b]furan-7-yl octanoate](http://img.cochemist.com/ccimg/67600/67526-95-8_b.png)


![Ethanone,1-[4-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]phenyl]-2,2,2-trifluoro-,O-[(4-methylphenyl)sulfonyl]oxime](http://img.cochemist.com/ccimg/87800/87736-80-9.png)
![Ethanone,1-[4-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]phenyl]-2,2,2-trifluoro-,O-[(4-methylphenyl)sulfonyl]oxime](http://img.cochemist.com/ccimg/87800/87736-80-9_b.png)
![Benzenemethanol, 4-[3-(trifluoromethyl)-3H-diazirin-3-yl]-](http://img.cochemist.com/ccimg/87800/87736-88-7.png)
![Benzenemethanol, 4-[3-(trifluoromethyl)-3H-diazirin-3-yl]-](http://img.cochemist.com/ccimg/87800/87736-88-7_b.png)


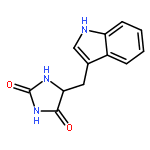
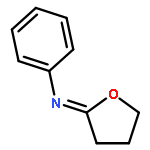
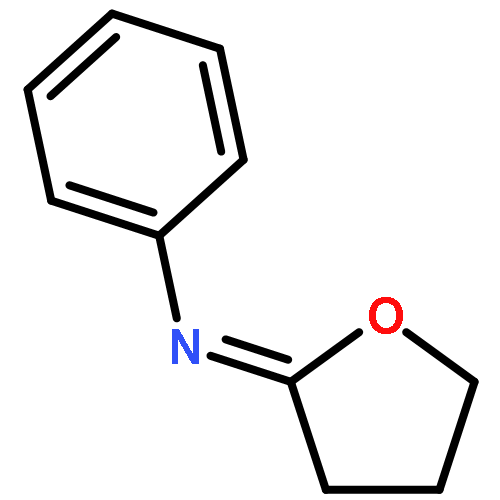


![5-Isoquinolinesulfonicacid,4-[(2S)-2-[(5-isoquinolinylsulfonyl)methylamino]-3-oxo-3-(4-phenyl-1-piperazinyl)propyl]phenylester](http://img.cochemist.com/ccimg/127200/127191-97-3.png)
![5-Isoquinolinesulfonicacid,4-[(2S)-2-[(5-isoquinolinylsulfonyl)methylamino]-3-oxo-3-(4-phenyl-1-piperazinyl)propyl]phenylester](http://img.cochemist.com/ccimg/127200/127191-97-3_b.png)
![Diaziridine, 3-[4-(methylthio)phenyl]-3-(trifluoromethyl)-](http://img.cochemist.com/ccimg/166900/166831-76-1.png)
![Diaziridine, 3-[4-(methylthio)phenyl]-3-(trifluoromethyl)-](http://img.cochemist.com/ccimg/166900/166831-76-1_b.png)
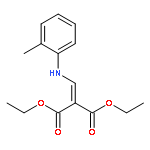
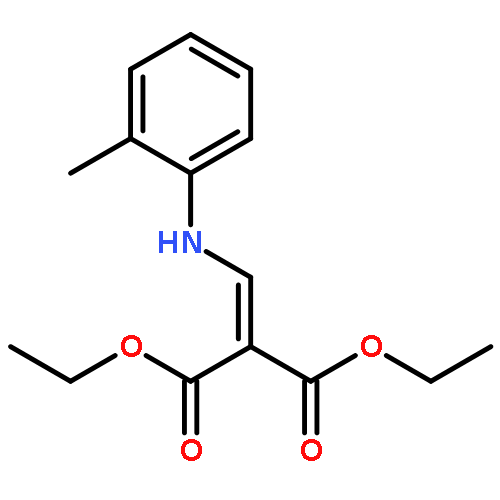


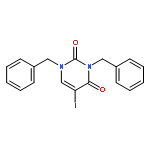
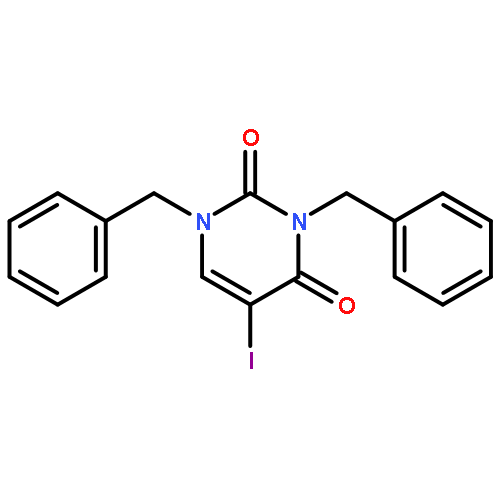
![Ethanone,2,2,2-trifluoro-1-[4-(methylthio)phenyl]-](http://img.cochemist.com/ccimg/122300/122243-33-8.png)
![Ethanone,2,2,2-trifluoro-1-[4-(methylthio)phenyl]-](http://img.cochemist.com/ccimg/122300/122243-33-8_b.png)
![1-Oxa-6-azaspiro[4.4]nonane-8-carbonitrile, 6-methyl-, (5R,8R)-rel-](http://img.cochemist.com/ccimg/127200/127144-42-7.png)
![1-Oxa-6-azaspiro[4.4]nonane-8-carbonitrile, 6-methyl-, (5R,8R)-rel-](http://img.cochemist.com/ccimg/127200/127144-42-7_b.png)
![[1,2,4]Triazolo[1,5-a]pyrimidine, 7-chloro-5-methyl-2-[(phenylmethyl)thio]-](/data/chemimg/1259300/51646-34-5.png)
![[1,2,4]Triazolo[1,5-a]pyrimidine, 7-chloro-5-methyl-2-[(phenylmethyl)thio]-](/data/chemimg/1259300/51646-34-5_b.png)
![Ethanone,1-[4-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]phenyl]-2,2,2-trifluoro-,oxime](http://img.cochemist.com/ccimg/87800/87736-77-4.png)
![Ethanone,1-[4-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]phenyl]-2,2,2-trifluoro-,oxime](http://img.cochemist.com/ccimg/87800/87736-77-4_b.png)
![[1,1'-Biphenyl]-4-aceticacid, methyl ester](http://img.cochemist.com/ccimg/59800/59793-29-2.png)
![[1,1'-Biphenyl]-4-aceticacid, methyl ester](http://img.cochemist.com/ccimg/59800/59793-29-2_b.png)
![[1,1'-Biphenyl]aceticacid](http://img.cochemist.com/ccimg/51400/51317-25-0.png)
![[1,1'-Biphenyl]aceticacid](http://img.cochemist.com/ccimg/51400/51317-25-0_b.png)
![1H-Benzimidazole, 1-[(3-chlorophenyl)methyl]-5,6-dimethyl-](http://img.cochemist.com/ccimg/141300/141211-38-3.png)
![1H-Benzimidazole, 1-[(3-chlorophenyl)methyl]-5,6-dimethyl-](http://img.cochemist.com/ccimg/141300/141211-38-3_b.png)


![4-[3-(Trifluoromethyl)-3H-diaziridine]benzyl Alcohol tert-Butyl(dimethyl)silyl Ether](http://img.cochemist.com/ccimg/87800/87736-83-2.png)
![4-[3-(Trifluoromethyl)-3H-diaziridine]benzyl Alcohol tert-Butyl(dimethyl)silyl Ether](http://img.cochemist.com/ccimg/87800/87736-83-2_b.png)
![6-Oxa-1-azaspiro[4.5]decane-3-carbonitrile, 1-methyl-, (3R,5R)-rel-](/data/chemimg/1075100/127144-44-9.png)
![6-Oxa-1-azaspiro[4.5]decane-3-carbonitrile, 1-methyl-, (3R,5R)-rel-](/data/chemimg/1075100/127144-44-9_b.png)
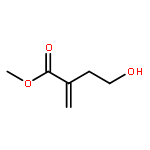
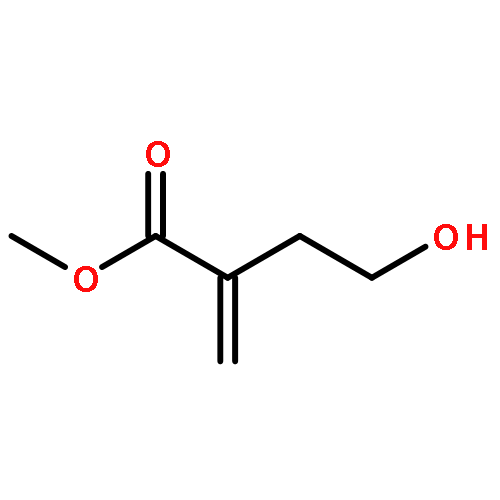
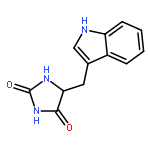

![2-Oxiranecarboxamide,3-[(4E,7E)-1-oxo-4,7-nonadien-1-yl]-, (2R,3S)-](http://img.cochemist.com/ccimg/17400/17397-89-6.png)
![2-Oxiranecarboxamide,3-[(4E,7E)-1-oxo-4,7-nonadien-1-yl]-, (2R,3S)-](http://img.cochemist.com/ccimg/17400/17397-89-6_b.png)


![Propanedioic acid,2-[[(4-methylphenyl)amino]methylene]-, 1,3-diethyl ester](http://img.cochemist.com/ccimg/19100/19056-84-9.png)
![Propanedioic acid,2-[[(4-methylphenyl)amino]methylene]-, 1,3-diethyl ester](http://img.cochemist.com/ccimg/19100/19056-84-9_b.png)
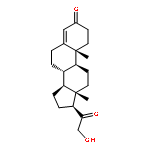
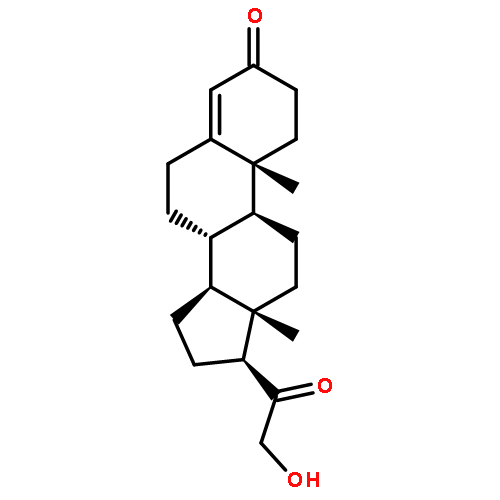
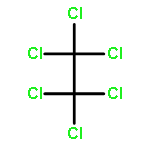
![Ethanone, 2,2,2-trifluoro-1-[3-(methylthio)phenyl]- (9CI)](http://img.cochemist.com/ccimg/166900/166831-66-9.png)
![Ethanone, 2,2,2-trifluoro-1-[3-(methylthio)phenyl]- (9CI)](http://img.cochemist.com/ccimg/166900/166831-66-9_b.png)
![Ethanone,2,2,2-trifluoro-1-[2-(methylthio)phenyl]-](http://img.cochemist.com/ccimg/166900/166831-65-8.png)
![Ethanone,2,2,2-trifluoro-1-[2-(methylthio)phenyl]-](http://img.cochemist.com/ccimg/166900/166831-65-8_b.png)
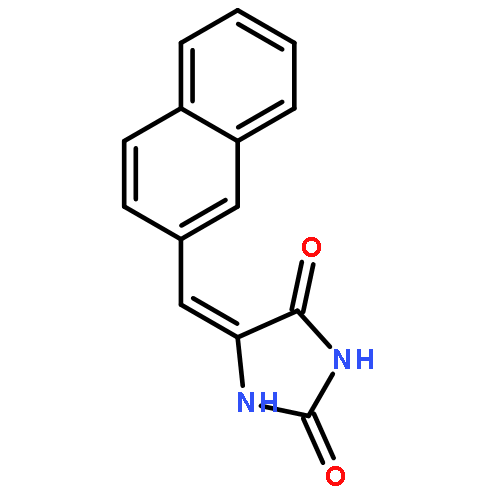
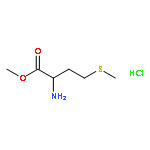
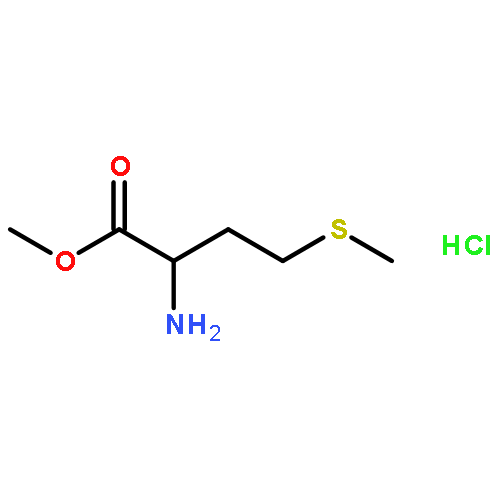


![6-Fluoro-2-(2'-fluoro-[1,1'-biphenyl]-4-yl)-3-methylquinoline-4-carboxylic acid](http://img.cochemist.com/ccimg/96200/96187-53-0.png)
![6-Fluoro-2-(2'-fluoro-[1,1'-biphenyl]-4-yl)-3-methylquinoline-4-carboxylic acid](http://img.cochemist.com/ccimg/96200/96187-53-0_b.png)
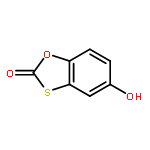
![Carbamic acid,N-[(1E,5R)-5-[3-[(2E,4E)-2,5-dimethyl-1-oxo-2,4-nonadien-1-yl]-4-hydroxy-2-oxo-2H-pyran-6-yl]-1-hexen-1-yl]-,methyl ester](http://img.cochemist.com/ccimg/88200/88192-99-8.png)
![Carbamic acid,N-[(1E,5R)-5-[3-[(2E,4E)-2,5-dimethyl-1-oxo-2,4-nonadien-1-yl]-4-hydroxy-2-oxo-2H-pyran-6-yl]-1-hexen-1-yl]-,methyl ester](http://img.cochemist.com/ccimg/88200/88192-99-8_b.png)



![Acetamide,N-(4,5-dihydro-4-methyl-5-oxo-1,2-dithiolo[4,3-b]pyrrol-6-yl)-](http://img.cochemist.com/ccimg/100/87-11-6.png)
![Acetamide,N-(4,5-dihydro-4-methyl-5-oxo-1,2-dithiolo[4,3-b]pyrrol-6-yl)-](http://img.cochemist.com/ccimg/100/87-11-6_b.png)

![2-PYRROLIDINEACETAMIDE, 4-[6-(1,4-DIMETHYLSPIRO[2,9-DIOXABICYCLO[3.3.1]NON-6-ENE-8,2'-OXIRAN-3-YL)-1-HYDROXY-4-METHYL-2,4-HEPTADIENYLIDENE]-N,.ALPHA.-DIMETHYL-3,5-DIOXO-1-(TETRAHYDRO-5-HYDROXY-6-METHYL-2H-PYRAN-2-YL)-](http://img.cochemist.com/ccimg/7300/7229-50-7.png)
![2-PYRROLIDINEACETAMIDE, 4-[6-(1,4-DIMETHYLSPIRO[2,9-DIOXABICYCLO[3.3.1]NON-6-ENE-8,2'-OXIRAN-3-YL)-1-HYDROXY-4-METHYL-2,4-HEPTADIENYLIDENE]-N,.ALPHA.-DIMETHYL-3,5-DIOXO-1-(TETRAHYDRO-5-HYDROXY-6-METHYL-2H-PYRAN-2-YL)-](http://img.cochemist.com/ccimg/7300/7229-50-7_b.png)






![(E)-6-[3-(3-CYCLOHEXYL-3-HYDROXYPROPYL)-2,5-DIOXOIMIDAZOLIDIN-4-YL]HEX-4-ENOIC ACID](http://img.cochemist.com/ccimg/78900/78864-51-4.png)
![(E)-6-[3-(3-CYCLOHEXYL-3-HYDROXYPROPYL)-2,5-DIOXOIMIDAZOLIDIN-4-YL]HEX-4-ENOIC ACID](http://img.cochemist.com/ccimg/78900/78864-51-4_b.png)












![4-Thiazolidinone, 5-[(5-nitro-2-furanyl)methylene]-2-thioxo-](http://img.cochemist.com/ccimg/13500/13410-84-9.png)
![4-Thiazolidinone, 5-[(5-nitro-2-furanyl)methylene]-2-thioxo-](http://img.cochemist.com/ccimg/13500/13410-84-9_b.png)
![8-FLUORO-2,3,4,5-TETRAHYDRO-1H-PYRIDO[4,3-B]INDOLE](http://img.cochemist.com/ccimg/39900/39876-39-6.png)
![8-FLUORO-2,3,4,5-TETRAHYDRO-1H-PYRIDO[4,3-B]INDOLE](http://img.cochemist.com/ccimg/39900/39876-39-6_b.png)
![N-(5-oxo-4,5-dihydro[1,2]dithiolo[4,3-b]pyrrol-6-yl)acetamide](http://img.cochemist.com/ccimg/500/488-04-0.png)
![N-(5-oxo-4,5-dihydro[1,2]dithiolo[4,3-b]pyrrol-6-yl)acetamide](http://img.cochemist.com/ccimg/500/488-04-0_b.png)





![3-[(4-CHLOROBENZYL)SULFANYL]-1H-1,2,4-TRIAZOL-5-YLAMINE](http://img.cochemist.com/ccimg/41300/41266-78-8.png)
![3-[(4-CHLOROBENZYL)SULFANYL]-1H-1,2,4-TRIAZOL-5-YLAMINE](http://img.cochemist.com/ccimg/41300/41266-78-8_b.png)










![3-Piperidinamine,N-[(2-methoxyphenyl)methyl]-2-phenyl-, (2S,3S)-](http://img.cochemist.com/ccimg/137000/136982-36-0.png)
![3-Piperidinamine,N-[(2-methoxyphenyl)methyl]-2-phenyl-, (2S,3S)-](http://img.cochemist.com/ccimg/137000/136982-36-0_b.png)
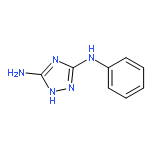

![[1,1'-Biphenyl]-4-amine,2'-chloro-](http://img.cochemist.com/ccimg/1300/1204-42-8.png)
![[1,1'-Biphenyl]-4-amine,2'-chloro-](http://img.cochemist.com/ccimg/1300/1204-42-8_b.png)


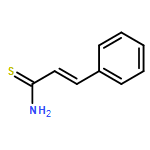
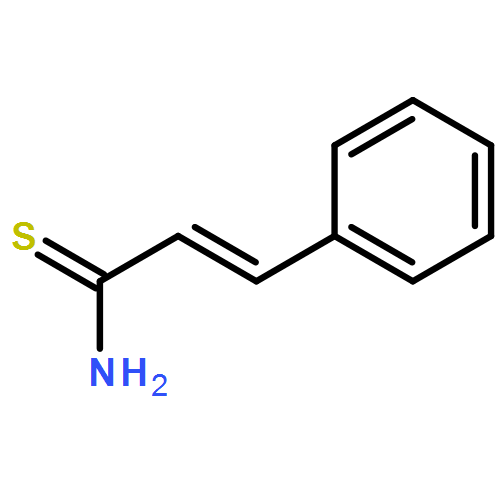






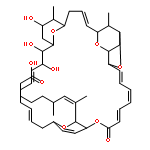



![Spiro[indene-1,4'-piperidine]](http://img.cochemist.com/ccimg/33100/33042-66-9.png)
![Spiro[indene-1,4'-piperidine]](http://img.cochemist.com/ccimg/33100/33042-66-9_b.png)




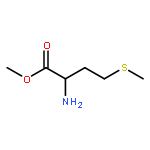

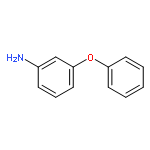

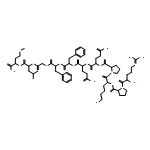
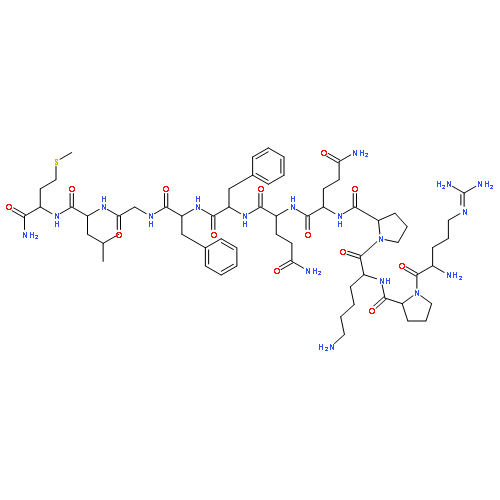






![4-(4-Fluorophenyl)-2-[4-(methylsulfinyl)phenyl]-5-(4-pyridyl)-1H-imidazole](http://img.cochemist.com/ccimg/152200/152121-47-6.png)
![4-(4-Fluorophenyl)-2-[4-(methylsulfinyl)phenyl]-5-(4-pyridyl)-1H-imidazole](http://img.cochemist.com/ccimg/152200/152121-47-6_b.png)












![2-[4-(benzyloxy)benzylidene]-N-(pyridin-3-yl)hydrazinecarbothioamide](http://img.cochemist.com/ccimg/5500/5453-51-0.png)
![2-[4-(benzyloxy)benzylidene]-N-(pyridin-3-yl)hydrazinecarbothioamide](http://img.cochemist.com/ccimg/5500/5453-51-0_b.png)