Co-reporter:Robert D. Bach
The Journal of Physical Chemistry A 2016 Volume 120(Issue 5) pp:840-850
Publication Date(Web):January 19, 2016
DOI:10.1021/acs.jpca.5b12086
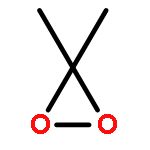
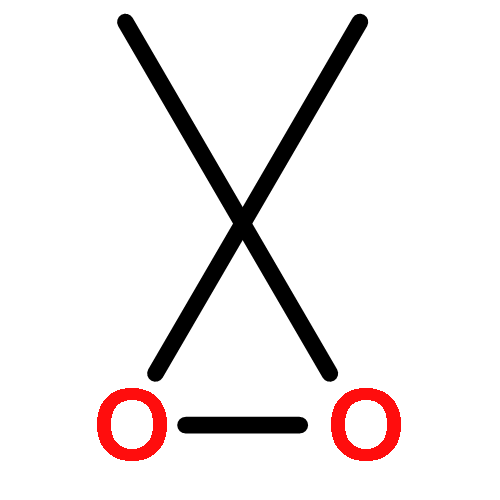
Both DFT and G4 molecular orbital calculations have been employed to reexamine the mechanism of dimethyldioxirane (DMDO) oxidation of saturated hydrocarbons and the epoxidation of alkenes. The UM062X DFT functional provided the most accurate bond O–O dissociation energies for a series of typical peroxides. A diradicaloid process initiated by an O–O homolytic bond cleavage involving abstraction of hydrogen from the C–H bond followed by a final product forming “oxygen rebound” step best describes the DMDO oxidation of saturated hydrocarbons. In contrast, this study showed that the DMDO epoxidation of alkenes is a concerted process best described with the B3LYP DFT functional.
Co-reporter:Robert D. Bach and Andrea Mattevi
The Journal of Organic Chemistry 2013 Volume 78(Issue 17) pp:8585-8593
Publication Date(Web):July 29, 2013
DOI:10.1021/jo401274u
DFT calculations presented for C(4a)-hydroperoxyflavin (C(4a)-FLHOOH) at the B3LYP/6-311+G(d,p) level suggest a new mechanism for the elimination of H2O2. The calculated activation barrier for a concerted four-centered elimination (ΔE‡ = 32.86 kcal/mol) strongly suggests that in the absence of interactions with the local environment a spontaneous elimination is not feasible. A proton shuttle from the N5 hydrogen to the proximal oxygen of the OOH moiety involving three water molecules has an activation barrier that is reduced to 17.11 kcal/mol. Calculations that utilize CH3OH to model the role of a local Thr or Ser residue shows that an alcohol functionality hydrogen bonded to the N5 H-atom can catalyze the elimination of H2O2 with a free energy of activation of 21.5 kcal/mol. Interaction of amines and amide residues (CH3NH2 and CH3(C═O)NH2) with the N5 locus of C(4a)-hydroperoxyflavin markedly reduce the activation barrier for H2O2 elimination relative to the concerted pathway. Proton transfer from a COOH group (ΔG‡ = 8.36 kcal/mol) or the NH2 group of a positively charged Arg model (ΔG‡ = 9.99 kcal/mol) to the proximal oxygen of the OOH moiety of C(4a)-FLHOOH in the TS for H2O2 elimination strongly enhances elimination of H2O2.
Co-reporter:Robert D. Bach
The Journal of Organic Chemistry 2012 Volume 77(Issue 16) pp:6801-6815
Publication Date(Web):July 31, 2012
DOI:10.1021/jo300727w
Quantum mechanical calculations at the B3LYP/6-311+G(d,p) level have examined the overall mechanism of the Baeyer–Villiger (BV) reaction with peroxyacetic acid. A series of reactions that include both the addition step and the subsequent alkyl group migration step included ketones, acetone, t-butyl methyl ketone, acetophenone, cyclohexyl methyl ketone, and cyclohexyl phenyl ketone. The combined data suggested that the first step for addition of the peroxyacetic acid oxidation catalyst to the ketone carbonyl to produce the Criegee or tetrahedral intermediate is rate-limiting and has activation barriers that range from 38 to 41 kcal/mol without the aid of a catalyst. The rate of addition is markedly reduced by the catalytic action of a COOH functionality acting as a donor–acceptor group affecting both its proton transfer to the ketone C═O oxygen in concert with transfer of the OOH proton to the carboxylic acid carbonyl. The second or alkyl group migration step has a much reduced activation barrier, and its rate is not markedly influenced by acid catalysis. The rate of both steps in the BV reaction is greatly influenced by the catalytic action of very strong acids.
Co-reporter:Robert D. Bach
The Journal of Physical Chemistry A 2011 Volume 115(Issue 40) pp:11087-11100
Publication Date(Web):September 2, 2011
DOI:10.1021/jp208087u
Model quantum mechanical calculations presented for C-4a-flavin hydroperoxide (FlHOOH) at the B3LYP/6-311+G(d,p) level suggest a new mechanism for flavoprotein monooxygenase (FMO) oxidation involving a concerted homolytic O–O bond cleavage in concert with hydroxyl radical transfer from the flavin hydroperoxide rather than an SN2-like displacement by the substrate on the C-4a-hydroperoxide OOH group. Homolytic O–O bond cleavage in a somersault-like rearrangement of hydroperoxide C-4a-flavinhydroperoxide (1) (FLHO–OH → FLHO···HO) produces an internally hydrogen-bonded HO• radical intermediate with a classical activation barrier of 27.0 kcal/mol. Model hydroperoxide 1 is used to describe the transition state for the key oxidation step in the paradigm aromatic hydroxylase, p-hydroxybenzoate hydroxylase (PHBH). A comparison of the electron distribution in the transition structures for the PHBH hydroxylation of p-hydroxybenzoic acid (ΔE‡ = 23.0 kcal/mol) with that of oxidation of trimethylamine (ΔE‡ = 22.3 kcal/mol) and dimethyl sulfide (ΔE‡ = 14.1 kcal/mol) also suggests a mechanism involving a somersault mechanism in concert with transfer of an HO• radical to the nucleophilic heteroatom center with a hydrogen transfer back to the FLH–O residue after the barrier is crossed to produce the final product, FLH–OH. In each case the hydroxylation barrier was less than that of the O–O rearrangement barrier in the absence of a substrate supporting an overall concerted process. All three transition structures bear a resemblance to the TS for the comparable hydroxylation of isobutane (ΔE‡ = 29.2 kcal/mol) and for simple Fenton oxidation by aqueous iron(III) hydroperoxides. To our surprise the oxidation of N- and S-nucleophiles with conventional oxidants such as alkyl hydroperoxides and peracids also proceeds by HO• radical transfer in a manner quite similar to that for tricyclic hydroperoxide 1. Stabilization of the developing oxyradical produced by somersault rearrangement for concerted enzymatic oxidation with tricyclic hydroperoxide 1 results in a reduced overall activation barrier.
Co-reporter:Robert D. Bach and Olga Dmitrenko
The Journal of Organic Chemistry 2010 Volume 75(Issue 11) pp:3705-3714
Publication Date(Web):April 30, 2010
DOI:10.1021/jo1004668
Quantum mechanical calculations (DFT) have provided a mechanism for the oxidative C−H bond cleavage step in Fenton-like hydrocarbon hydroxylation. A transition structure for hydrocarbon oxidation by aqueous solvated cationic iron(III) hydroperoxides ((H2O)nFeIIIOOH) is presented that involves a novel rearrangement of the hydroperoxide group (FeO−OH → FeO···HO) in concert with hydrogen abstraction by the incipient HO• radical with activation barriers ranging from 17 to 18 kcal/mol. In every hydroperoxide examined, the activation barrier for FeO−OH isomerization, in the absence of the hydrocarbon, is significantly greater than the overall concerted activation barrier for C−H bond cleavage in support of the concept of O−O bond isomerization in concert with hydrogen abstraction. The transition structure for the oxidation step in simple anionic iron(III) hydroperoxides has been shown to bear a remarkable resemblance to model porphyrin calculations on cytochrome P450 hydroxylation.
Co-reporter:Robert D. Bach
The Journal of Physical Chemistry A 2010 Volume 114(Issue 34) pp:9319-9332
Publication Date(Web):August 6, 2010
DOI:10.1021/jp1045518
Model theoretical quantum mechanical (QM) calculations are described for the P-450 hydroxylation of methane, isobutane, and camphor that compare the concerted somersault H-abstraction mechanism with the oxidation step involving Cpd I. Special emphasis has been placed on maintaining a balanced basis set in the oxidation step. QM calculations, employing the 6-311+G(d,p) basis set on the Fe atom and all of the key surrounding atoms involved in the C−H abstraction step, reaffirm a mechanism involving rearrangement of the iron hydroperoxide group (FeO−OH → FeO···HO•) in concert with hydrogen abstraction from the C−H bond of the substrate by the incipient bound hydroxyl radical HO•. The barrier for the somersault rearrangement of model Cpd 0 (FeO−OH) is calculated to be 21.4 kcal/mol in the absence of substrate. The overall activation energy for the oxidation of camphor involving the somersault motion of the FeO−OH group of P450 model porphyrin iron(III) hydroperoxide [Por(SH)Fe(III)−OOH−] → [Por(SH)Fe(III)−O····HO−] in concert with hydrogen abstraction is ΔE‡ = 12.4 kcal/mol. The corresponding abstraction of the hydrogen atom from the C−H bond of camphor by Cpd I has an activation barrier of 17.6 kcal/mol. Arguments are presented that the somersault rearrangement is induced by steric compression at the active site. Kinetic isotope effect data are discussed that provides compelling evidence for a rate-limiting step involving C−H bond cleavage.