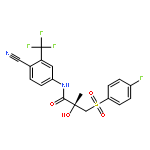
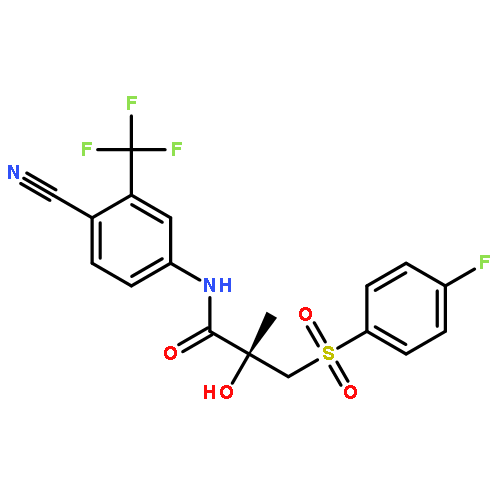
Co-reporter:Ling Zhang, Dian He, Kun Li, Hongli Liu, Baitao Wang, Lifang Zheng, Jiazhong Li
Biomedicine & Pharmacotherapy 2017 Volume 90(Volume 90) pp:
Publication Date(Web):1 June 2017
DOI:10.1016/j.biopha.2017.03.046
•Emodin localized in mitochondria analyzed by its inherent fluorescence.•CypD was involved in the apoptosis induced by emodin.•ROS and ERK played a role in the regulation of CypD.•Molecule docking disclosed three hydrogen bonds existed in the emodin-CypD complex.Emodin has demonstrated potent anticancer activity in human hepatocarcinoma cells and animal models, however, the cellular targets of emodin have not been fully defined. Here we report that emodin induces the dysfunction of mitochondria and the apoptosis in HepG2 cells through an enrichment in mitochondria. Specifically, A mitochondrial matrix protein (cyclophilin D, CyPD) is involved in emodin-induced apoptosis, and the inhibitor of CyPD (cyclosporin A) could almost completely suppressing the apoptosis; Moreover, as the expression of CyPD could be effectively inhibited by antioxidant N-acetyl-l-cysteine and epidermal growth factor (the activator of ERK), reactive oxygen species and ERK might be involved in the relevant role of CyPD. A further molecule-docking discloses the existence of three hydrogen-bonds in CyPD-emodin complex. Thus, target localization and CyPD in mitochondria provides an insight into the action of emodin in the treatment of liver cancer.Download high-res image (103KB)Download full-size image
Co-reporter:Hongli Liu;Rui Han
Journal of Computer-Aided Molecular Design 2016 Volume 30( Issue 12) pp:1189-1200
Publication Date(Web):2016 December
DOI:10.1007/s10822-016-9992-2
R-bicalutamide, a first generation antiandrogen, was used to treat prostate cancer for decades. Although it is very effective at the beginning, resistance appears after 2–3 years of treatment. Mutation of androgen receptor (AR) is considered a main reason for drug resistance. It is reported that AR W741C, W741L, W741C_T877A, T877A, F876L, F876L_T877A and L701H mutations can convert R-bicalutamide from AR antagonist to agonist, but the switching mechanisms are not clear. In this study, molecular dynamics simulations and molecular mechanics generalized Born surface area (MM-GBSA) calculations were performed to analyze the interaction mechanisms between R-bicalutamide and wild type/mutant ARs. The results indicate that helix H12, which lies on the top of AR LBD like a cover, plays a vital role in R-bicalutamide binding. When interacting with AR, the B-ring of R-bicalutamide pushes H12 aside, distorting the coactivator binding site (AF2) resulting in the inactivation of transcription. Several residue mutations appear to enlarge the distance between the B-ring of R-bicalutamide and H12, reducing steric clash, which is conducive to a closed H12 conformation, leading to the formation of the coactivator binding site AF2 and increased transcription. Hydrogen bond and per-residue free energy decomposition analyses are also investigated to explore the interacting mechanisms, and M895 is found to be a key residue in the antagonist mechanism. The obtained molecular mechanisms will aid rational screening and design of novel AR antagonists, even to mutant AR.
Co-reporter:Jiazhong Li;Fang Bai;Huanxiang Liu;Paola Gramatica
Chemical Biology & Drug Design 2015 Volume 86( Issue 6) pp:1501-1517
Publication Date(Web):
DOI:10.1111/cbdd.12619
The concept of ligand efficiency is defined as biological activity in each molecular size and is widely accepted throughout the drug design community. Among different LE indices, surface efficiency index (SEI) was reported to be the best one in support vector machine modeling, much better than the generally and traditionally used end-point pIC50. In this study, 2D multiple linear regression and 3D comparative molecular field analysis methods are employed to investigate the structure–activity relationships of a series of androgen receptor antagonists, using pIC50 and SEI as dependent variables to verify the influence of using different kinds of end-points. The obtained results suggest that SEI outperforms pIC50 on both MLR and CoMFA models with higher stability and predictive ability. After analyzing the characteristics of the two dependent variables SEI and pIC50, we deduce that the superiority of SEI maybe lie in that SEI could reflect the relationship between molecular structures and corresponding bioactivities, in nature, better than pIC50. This study indicates that SEI could be a more rational parameter to be optimized in the drug discovery process than pIC50.
Co-reporter:Hongli Liu, Xiaoli An, Shuyan Li, Yuwei Wang, Jiazhong Li and Huanxiang Liu
Molecular BioSystems 2015 vol. 11(Issue 12) pp:3347-3354
Publication Date(Web):25 Sep 2015
DOI:10.1039/C5MB00499C
R-Bicalutamide is a first generation antiandrogen used to treat prostate cancer, which inhibits androgen action by competitively binding to the androgen receptor (AR). However, R-bicalutamide was discovered to exhibit some agonistic properties in clinical application. According to reports, the W741L AR mutation may lead to resistance towards R-bicalutamide. But the mechanism of the R-bicalutamide switch from an antagonist to an agonist due to the mutation of AR W741L is still not so clear. Another molecule, S-1, owing to a very similar structure to R-bicalutamide, is always agonistic to both the wild type and W741L AR. The main difference between these two chemicals is that S-1 has an ether linkage while R-bicalutamide has a sulfonyl group. To study the drug-resistant mechanism caused by W741L mutation and the opposite effects arising from subtle structure differences, molecular dynamics (MD) simulations and molecular mechanics generalized Born surface area (MM-GBSA) calculations were employed to explore the interaction mechanisms between R-bicalutamide/S-1 and WT/W741L AR. The calculated binding free energies are in accordance with the reported experimental values. The obtained results indicate that M895 and W741 are vital amino acids in the antagonism of R-bicalutamide. The bulkier substitution of sulfonyl and tryptophan push aside M895, together with helix 12 (H12), to expose the ligand-binding domain resulting in the antagonistic conformation of the AR. If W741 is mutated to L741, the B-ring of these two chemicals would shift toward L741. At the same time, M895 dragging helix H12, would also move closer to L741. So H12 tends to cover the AR ligand-binding domain to a certain degree, changing the androgen receptor from an antagonistic to an agonistic conformation, which may explain the agonism of R-bicalutamide to the mutant W741L AR.
Co-reporter:Jiazhong Li, Shuyan Li, Chongliang Bai, Huanxiang Liu, Paola Gramatica
Journal of Molecular Graphics and Modelling 2013 Volume 44() pp:266-277
Publication Date(Web):July 2013
DOI:10.1016/j.jmgm.2013.07.004
•Robust and predictive models were built by using MLR, CoMFA and CoMSIA methods.•Position R1: A small hydrophobic electro-donating group benefits the bioactivity.•Position R2: A hydrophobic and electronegative atom is expected, especially halogen.•Position R4: Important to the solubility, an electro-withdrawing group is expected.•Position R5: A bulky connected with benzene by an electro-donating atom is preferred.Malaria is a fatal tropical and subtropical disease caused by the protozoal species Plasmodium. Many commonly available antimalarial drugs and therapies are becoming ineffective because of the emergence of multidrug resistant Plasmodium falciparum, which drives the need for the development of new antimalarial drugs. Recently, a series of 3-carboxyl-4(1H)-quinolone analogs, derived from the famous compound endochin, were reported as promising candidates for orally efficacious antimalarials. In this study, to analyze the structure–activity relationships (SAR) of these quinolones and investigate the structural requirements for antimalarial activity, the 2D multiple linear regressions (MLR) method and 3D comparative molecular field analysis (CoMFA) and comparative molecular similarity indices analysis (CoMSIA) methods are employed to evolve different QSAR models. All these models give satisfactory results with highly accurate fitting and strong external predictive abilities for chemicals not used in model development. Furthermore, the contour maps from 3D models can provide an intuitive understanding of the key structure features responsible for the antimalarial activities. In conclusion, we summarize the detailed position-specific structural requirements of these derivatives accordingly. All these results are helpful for the rational design of new compounds with higher antimalarial bioactivities.Graphical abstract
Co-reporter:Xing Huo, Jiazhong Li, Paola Gramatica
Chemometrics and Intelligent Laboratory Systems 2011 Volume 107(Issue 2) pp:283-289
Publication Date(Web):July 2011
DOI:10.1016/j.chemolab.2011.04.013
In the clinical treatment of prostate cancer, mutation in the androgen receptor (AR) that occurred in patients, often results in drug resistance because the agonist activity of the drug increases while the antagonist activity decreases. Recently a novel series of diazacycloundecane AR antagonists were reported, and their abilities to antagonize the wild type (WT) AR as well as antagonize the mutated type (MT) AR (T877A) were determined experimentally. In this paper, to understand more about the drug resistance and to help to design more active antagonists, the quantitative structure–activity relationship (QSAR) of these compounds were analyzed, and the different interactions of these antagonists with two types of AR were compared. As a result two robust and predictive QSAR models were proposed, and the different structure features related with the antagonist activities on WT and MT AR were analyzed separately.

![5-NITRO-1-(PHENYLSULFONYL)-1H-PYRROLO[2,3-B]PYRIDINE-2-CARBALDEHYDE](http://img.cochemist.com/ccimg/458600/458559-34-7.png)
![5-NITRO-1-(PHENYLSULFONYL)-1H-PYRROLO[2,3-B]PYRIDINE-2-CARBALDEHYDE](http://img.cochemist.com/ccimg/458600/458559-34-7_b.png)






![(2-ISOPROPYL-3-INDOLIZINYL)(4-{3-[(2-METHYL-2-PROPANYL)AMINO]PROP<WBR />OXY}PHENYL)METHANONE](http://img.cochemist.com/ccimg/3600/3546-21-2.png)
![(2-ISOPROPYL-3-INDOLIZINYL)(4-{3-[(2-METHYL-2-PROPANYL)AMINO]PROP<WBR />OXY}PHENYL)METHANONE](http://img.cochemist.com/ccimg/3600/3546-21-2_b.png)